⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
Comprehensive Report on Lou Gehrig’s Disease (ALS)
1. Overview
What is Lou Gehrig’s Disease?
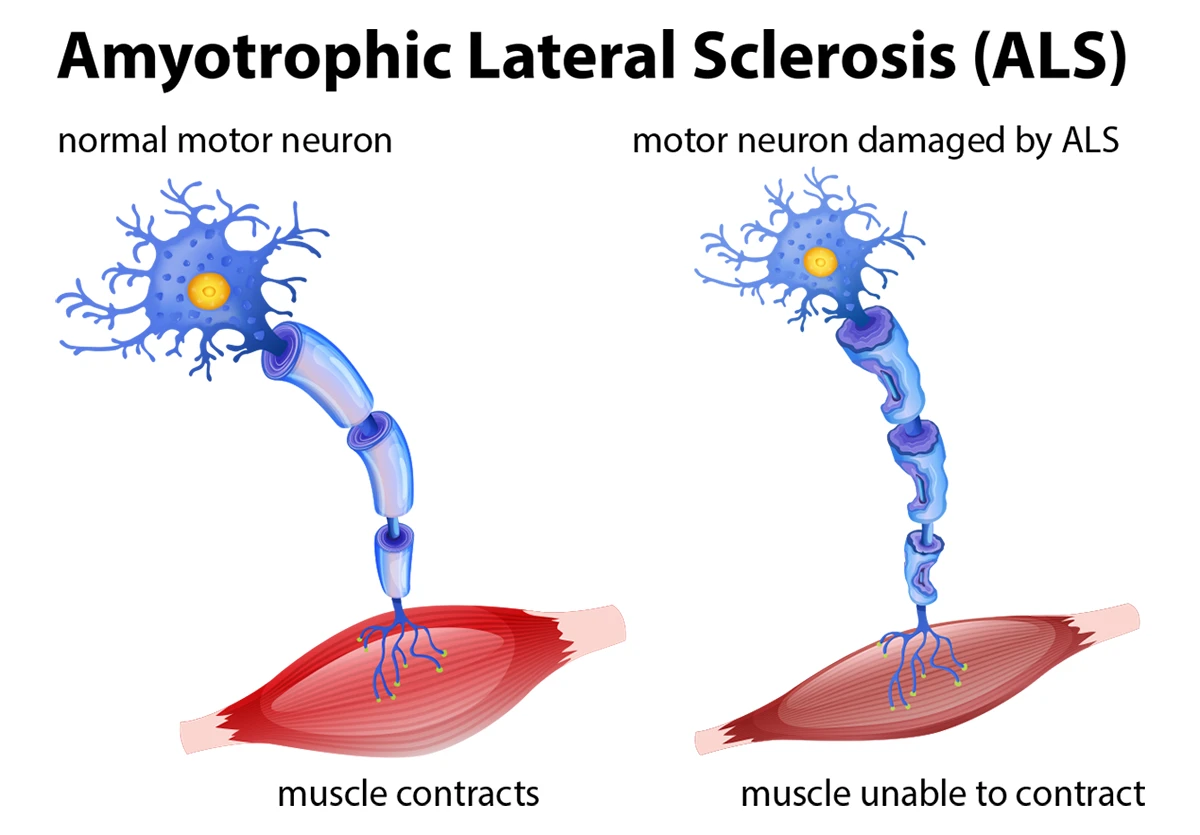
Lou Gehrig’s disease, formally known as Amyotrophic Lateral Sclerosis (ALS), is a progressive neurodegenerative disease that affects nerve cells in the brain and spinal cord. The name “Lou Gehrig’s disease” comes from the famous baseball player who was diagnosed with the condition in 1939, bringing national and worldwide attention to this previously little-known disorder.
Detailed Definition
ALS is characterized by the gradual degeneration and death of motor neurons—the nerve cells responsible for controlling voluntary muscle movement. Motor neurons extend from the brain to the spinal cord and from the spinal cord to muscles throughout the body. As these motor neurons deteriorate, they lose the ability to send signals to the muscles, leading to muscle weakness, atrophy, and eventually paralysis.
Affected Body Parts/Organs
ALS primarily affects:
- Upper motor neurons in the brain (particularly the motor cortex)
- Lower motor neurons in the brainstem and spinal cord
- Voluntary muscles throughout the body (including those in the arms, legs, diaphragm, and throat)
- Respiratory system (as the disease progresses)
Notably, ALS generally does not affect:
- Sensory neurons (touch, pain, temperature sensation remain intact)
- Autonomic nervous system (controlling involuntary functions)
- Cognitive function in most cases (though a subset of patients develop frontotemporal dementia)
- Bladder and bowel control (until very late stages)
- Eye muscles (preserved even in advanced stages)
Prevalence and Significance
- Incidence: Approximately 1-2 new cases per 100,000 population annually
- Prevalence: About 5-7 cases per 100,000 population worldwide
- In the United States: Approximately 16,000-20,000 people living with ALS at any given time
- Median age of onset: Late 50s, though the disease can occur at any adult age
- Male to female ratio: Historically 1.5:1, though the gap has been narrowing in recent decades
- Economic impact: Annual cost of care estimated at $50,000-$200,000 per patient
ALS is significant not only because of its devastating physical effects but also because it represents one of the most severe neurodegenerative conditions with no effective cure. Despite its relatively rare status, ALS has gained substantial public awareness through advocacy campaigns such as the “Ice Bucket Challenge” in 2014, which raised over $115 million for research.
2. History & Discoveries
First Identification
ALS was first described in medical literature in 1869 by the French neurologist Jean-Martin Charcot. Through careful clinical observation and subsequent autopsy studies, Charcot identified the relationship between the symptoms experienced by his patients and the physical changes in their spinal cords. He named the condition “amyotrophic lateral sclerosis”—a term that describes its key pathological features:
- “Amyotrophic” refers to muscle atrophy
- “Lateral” identifies the areas of the spinal cord affected
- “Sclerosis” describes the hardening of tissue that replaces normal structures
Who Discovered It?
Jean-Martin Charcot (1825-1893), often referred to as “the father of modern neurology,” is credited with the discovery and first comprehensive description of ALS. Charcot worked at the Salpêtrière Hospital in Paris, where he established the world’s first neurology clinic and developed systematic methods for correlating clinical symptoms with pathological findings.
Major Discoveries and Breakthroughs
- 1869: Charcot’s original clinical and pathological description
- 1939: Lou Gehrig’s diagnosis brought widespread public attention to the disease
- 1962: Discovery of Guamanian ALS, a high-incidence form of the disease observed in Guam
- 1993: Identification of the first ALS-linked gene, SOD1 (superoxide dismutase 1)
- 1995: FDA approval of riluzole, the first drug for ALS
- 2006: Discovery of TDP-43 protein aggregates in ALS patients’ neurons
- 2011: Identification of C9orf72 repeat expansions as the most common genetic cause of familial ALS
- 2017: FDA approval of edaravone (Radicava), the second drug for ALS
- 2018-2020: Identification of multiple new genetic risk factors through genome-wide association studies
- 2022: Phase 3 clinical trial results suggesting benefit from AMX0035 (Relyvrio), leading to FDA approval
Evolution of Medical Understanding
The understanding of ALS has evolved dramatically:
- Early period (1869-1950s): ALS was viewed primarily as a disorder of motor neurons with limited clinical recognition and no effective treatments
- Middle period (1960s-1990s): Recognition of different ALS phenotypes and the beginnings of understanding environmental risk factors
- Genetic era (1993-2010): Discovery of ALS-linked genes revealing the hereditary component of some cases
- Modern era (2010-present): Recognition of ALS as a heterogeneous, complex disorder involving:
- Protein misfolding and aggregation
- RNA processing abnormalities
- Mitochondrial dysfunction
- Neuroinflammation
- Glutamate excitotoxicity
- Axonal transport defects
- Connections to frontotemporal dementia
The disease is now understood to exist on a spectrum of neurodegenerative disorders rather than as a singular entity, with significant overlap with frontotemporal dementia and other neurodegenerative conditions.
3. Symptoms
Early Symptoms vs. Advanced-Stage Symptoms
Early Symptoms:
- Asymmetric limb weakness, typically beginning in one hand or foot
- Subtle muscle twitches (fasciculations)
- Muscle cramps, particularly during physical activity
- Mild stiffness (spasticity)
- Fatigue that seems excessive for activity level
- Slurred speech (dysarthria) in bulbar-onset cases
- Difficulty swallowing (dysphagia) in bulbar-onset cases
- Increased tendency to trip or stumble
- Decreased fine motor control (difficulty with buttons, writing)
- Emotional lability (inappropriate laughing or crying)
Advanced-Stage Symptoms:
- Widespread muscle paralysis
- Significant muscle atrophy
- Respiratory insufficiency requiring ventilation support
- Complete loss of speech
- Inability to swallow, requiring feeding tube (PEG) placement
- Immobility requiring full-time care
- Potential cognitive changes (in some patients)
- Eye movement control often preserved, allowing for communication devices
- Pain from immobility, joint contractures, or muscle cramps
- Weight loss and metabolic changes
Common vs. Rare Symptoms
Common Symptoms (>50% of patients):
- Progressive limb weakness
- Muscle atrophy
- Fasciculations (muscle twitches)
- Hyperreflexia (increased reflexes)
- Spasticity (muscle stiffness)
- Dysarthria (slurred speech)
- Dysphagia (difficulty swallowing)
- Respiratory insufficiency (as disease progresses)
- Fatigue
- Weight loss
Rare Symptoms (<10% of patients):
- Frontotemporal dementia
- Sensory abnormalities
- Autonomic dysfunction (blood pressure fluctuations, sweating abnormalities)
- Bladder/bowel dysfunction (in early stages)
- Parkinsonism-like features
- Pain as a primary symptom (though secondary pain is common)
- Urinary or fecal incontinence (until very late stages)
- Ocular motor abnormalities
- Pressure ulcers (preventable with proper care)
Symptom Progression Over Time
ALS typically progresses through several stages, though the rate varies considerably between individuals:
Stage 1 (Early):
- Localized weakness in one region (limb or bulbar muscles)
- Normal daily functioning with minor adaptations
- Duration: Typically 3-12 months
Stage 2 (Middle):
- Weakness spreading to additional body regions
- Noticeable impact on daily activities
- Beginning of speech or swallowing difficulties if not present initially
- Duration: Typically 12-24 months
Stage 3 (Advanced):
- Weakness in most muscle groups
- Significant disability requiring assistive devices
- Respiratory involvement beginning
- Communication challenges
- Duration: Typically 12-24 months
Stage 4 (Terminal):
- Profound weakness in all limbs
- Respiratory failure requiring ventilation decisions
- Complete dependence for all activities
- Communication often limited to eye-controlled devices
- Duration: Variable depending on ventilation decisions
The progression rate varies significantly:
- Approximately 10% of patients have very slow progression (>10 years survival)
- About 10% have very rapid progression (<1 year survival)
- The majority fall between these extremes, with typical survival of 3-5 years from symptom onset
4. Causes
Biological and Environmental Causes
Biological Mechanisms:
- Motor neuron degeneration due to multiple proposed mechanisms:
- Protein misfolding and aggregation (especially TDP-43, SOD1, FUS proteins)
- Glutamate excitotoxicity (excessive neuron stimulation)
- Oxidative stress (free radical damage)
- Mitochondrial dysfunction (cellular energy failure)
- Disrupted axonal transport (materials not moving properly within neurons)
- Neuroinflammation (immune system attacking neurons)
- Disrupted RNA processing (errors in protein production)
- Cytoskeletal abnormalities (structural problems in neurons)
Environmental Factors with Evidence:
- Military service (particularly Gulf War veterans)
- Exposure to certain pesticides and heavy metals
- Smoking (modest increase in risk)
- Physical trauma, particularly head injuries
- Intense physical exertion in vulnerable individuals (controversial)
- Exposure to cyanobacteria (blue-green algae)
- Electric shock exposure
- Exposure to electromagnetic fields (limited evidence)
Genetic and Hereditary Factors
- Familial ALS (FALS): Accounts for 5-10% of all ALS cases
- Sporadic ALS (SALS): 90-95% of cases, but many have genetic contributions
Major ALS-associated genes:
- C9orf72 (most common genetic cause in Western populations, 40% of FALS)
- SOD1 (superoxide dismutase 1, 20% of FALS)
- TARDBP (encodes TDP-43 protein, 5% of FALS)
- FUS (fused in sarcoma protein, 5% of FALS)
- Other genes: UBQLN2, VCP, OPTN, TBK1, TUBA4A, KIF5A, and others
Inheritance patterns:
- Autosomal dominant (most common)
- Autosomal recessive (less common)
- X-linked (rare)
- Complex inheritance with multiple risk alleles (increasingly recognized)
Known Triggers or Exposure Risks
- Viral infections (proposed but not conclusively proven)
- Traumatic events (physical injuries, surgeries)
- Periods of intense physical exertion
- Exposure to environmental toxins
- Periods of psychological stress (possibly triggering underlying vulnerability)
- Recent evidence suggests “cluster susceptibility” where multiple factors must converge
The current scientific consensus views ALS as a multifactorial disease requiring both genetic susceptibility and environmental triggers, with the specific combination varying between individuals.
5. Risk Factors
Who Is Most at Risk
- Age: Most common between 55-75 years, though can occur in 20s-30s
- Gender: Historically males at higher risk (1.5:1), though the gap has been narrowing
- Race/Ethnicity: Higher rates in Caucasians than in African, Asian, or Hispanic populations
- Family History: Individuals with first-degree relatives with ALS or frontotemporal dementia
- Military Service: Particularly Gulf War veterans (approximately 1.5-2× increased risk)
- Athletic Activity: Professional athletes in certain sports (controversial):
- Italian soccer players (6× higher risk in one study)
- NFL players (4× higher risk in several studies)
- Marathon runners (inconclusive evidence)
Environmental, Occupational, and Genetic Factors
Environmental Factors:
- Rural living (possibly related to environmental exposures)
- Exposure to agricultural chemicals
- Lead and other heavy metal exposure
- Smoking (modest 1.2× increased risk)
- Beta-methylamino-L-alanine (BMAA) exposure from cyanobacteria
Occupational Factors:
- Military service
- Professional athletics (certain sports)
- Electrical work
- Welding
- Agricultural work
- Veterinary medicine
- Healthcare professionals (modest increased risk in some studies)
Genetic Factors:
- Over 30 genes identified with ALS association
- Polygenic risk scores now being developed
- Some genetic variants more common in specific populations:
- C9orf72 expansions most common in European populations
- SOD1 mutations more evenly distributed globally
- Specific mutations in Korean and Japanese populations
Impact of Pre-existing Conditions
- Frontotemporal dementia: Strong overlap, shared genetic risk
- Other neurodegenerative disorders: Possible shared vulnerability
- Autoimmune disorders: Conflicting evidence, potential relationship
- Metabolic syndrome: Possible protective effect (controversial)
- Head trauma history: Increased risk, particularly with repeated injuries
- Thyroid disease: Modest association in some studies
- Chronic inflammatory conditions: Potential relationship being studied
Interestingly, some conditions appear protective:
- History of autoimmune disorders has shown conflicting results but may be protective in some cases
- Higher pre-diagnosis BMI associated with better prognosis
- Cancer history may be associated with lower ALS risk (reverse comorbidity)
6. Complications
Complications Arising from ALS
Primary Complications:
- Respiratory failure (leading cause of death)
- Aspiration pneumonia (from swallowing difficulties)
- Malnutrition and dehydration
- Falls and related injuries
- Pressure ulcers (bedsores)
- Deep vein thrombosis from immobility
- Depression and anxiety
- Communication challenges leading to social isolation
- Pseudobulbar affect (uncontrollable laughing or crying)
- Insomnia and sleep disruption
- Constipation from immobility and medication side effects
- Cognitive changes (in some patients)
Secondary Complications:
- Medication side effects
- Ventilator-associated complications (in those choosing mechanical ventilation)
- Feeding tube complications
- Urinary tract infections
- Contractures (permanent joint tightening)
- Psychological impact on family/caregivers
- Financial hardship from care costs
Long-term Impact on Organs and Overall Health
Respiratory System:
- Progressive weakening of diaphragm and accessory breathing muscles
- Reduced vital capacity and breathing reserves
- Eventual respiratory failure without ventilation support
- Carbon dioxide retention leading to headaches, confusion
Musculoskeletal System:
- Widespread muscle atrophy
- Joint contractures and deformities
- Bone density loss from immobility
Digestive System:
- Dysphagia leading to malnutrition and weight loss
- Aspiration risk affecting eating and drinking
- Constipation from immobility and medications
- Altered metabolism with disease progression
Cardiovascular System:
- Indirect effects from immobility
- Increased risk of blood clots
- Generally preserved cardiac function
Urinary System:
- Generally preserved function until late stages
- Increased risk of urinary tract infections with immobility
Nervous System:
- Progressive motor neuron loss
- Frontotemporal dementia in some cases (15-20%)
- Sensory systems typically spared
- Pain from secondary causes (immobility, spasticity)
Potential Disability or Fatality Rates
Disability Progression:
- 50% of patients require significant assistive devices within 2 years of diagnosis
- 80% have significant bulbar dysfunction by 5 years if they survive
- Nearly 100% require full assistance with activities of daily living in advanced stages
Mortality:
- Median survival: 3-5 years from symptom onset
- Approximately 20% survive 5+ years
- 10% survive 10+ years
- 2-5% may have survival of 20+ years (very slow progression)
Survival variability by onset type:
- Bulbar onset: 2-3 years median survival
- Limb onset: 3-5 years median survival
- Respiratory onset: <2 years median survival
Mortality causes:
- Respiratory failure: 70%
- Pneumonia: 20%
- Other complications: 10%
Ventilation decisions dramatically impact survival times, with invasive ventilation potentially extending life by many years, though with complete paralysis.
7. Diagnosis & Testing
Common Diagnostic Procedures
Clinical Evaluation:
- Detailed neurological examination
- Muscle strength testing
- Reflex assessment
- Muscle tone evaluation
- Bulbar function assessment (speech, swallowing)
- Respiratory function tests
El Escorial Criteria: The diagnosis of ALS is based on the revised El Escorial criteria, which require:
- Evidence of lower motor neuron degeneration (by clinical, electrophysiological, or neuropathological examination)
- Evidence of upper motor neuron degeneration (by clinical examination)
- Progressive spread of symptoms within a region or to other regions
- Absence of electrophysiological or pathological evidence of other disease processes
- Absence of neuroimaging evidence of other disease processes
Medical Tests
Electrophysiological Tests:
- Electromyography (EMG): Shows denervation and reinnervation patterns
- Nerve conduction studies (NCS): Typically normal sensory function with reduced motor responses
- Motor unit number estimation (MUNE): Quantifies motor neuron loss
- Transcranial magnetic stimulation: Assesses upper motor neuron function
Imaging Studies:
- MRI of brain and spinal cord: Primarily to exclude other conditions
- Advanced MRI techniques:
- Diffusion tensor imaging (DTI)
- Magnetic resonance spectroscopy (MRS)
- Functional MRI (fMRI)
- PET scanning: Research tool for metabolism assessment
Laboratory Tests:
- Complete blood count
- Comprehensive metabolic panel
- Creatine kinase (often mildly elevated)
- Thyroid function tests
- Serum protein electrophoresis
- Vitamin B12 and folate levels
- Erythrocyte sedimentation rate
- Anti-ganglioside antibodies
- Heavy metal screening (in selected cases)
- Paraneoplastic antibody panel
- HIV testing (in at-risk individuals)
Genetic Testing:
- Testing for SOD1, C9orf72, TARDBP, FUS mutations
- Expanded panels for less common mutations
- Whole genome or exome sequencing in research settings
Other Procedures:
- Muscle biopsy (occasionally)
- Cerebrospinal fluid analysis (to exclude inflammatory conditions)
- Respiratory function tests (spirometry, forced vital capacity)
- Swallowing studies (videofluoroscopy)
- Sleep studies (for respiratory involvement assessment)
Early Detection Methods and Their Effectiveness
Current Early Detection Approaches:
- Clinical vigilance for early symptoms
- Targeted genetic testing in familial cases
- Electromyography showing subclinical denervation
Effectiveness of Early Detection:
- Average diagnostic delay: 10-18 months from symptom onset
- False positive rate for initial ALS diagnosis: 5-10%
- False negative rate for initial presentation: 30-40% (diagnosed as something else first)
Emerging Early Detection Research:
- Neurofilament light chain (NfL) blood/CSF biomarkers
- MicroRNA profiles
- Proteomics approaches
- Skin biopsy for TDP-43 pathology
- Advanced neuroimaging techniques
- Voice analysis for pre-symptomatic bulbar changes
- Muscle ultrasound for fasciculation detection
Early detection remains challenging due to:
- Symptom overlap with more common conditions
- Gradual symptom onset
- Variable presentation patterns
- Lack of a single definitive test
- Psychological barriers to seeking evaluation for subtle symptoms
Despite these challenges, the average time to diagnosis has decreased in recent decades due to greater awareness and improved diagnostic approaches.
8. Treatment Options
Standard Treatment Protocols
Multidisciplinary Care: The foundation of ALS management is coordinated multidisciplinary care involving:
- Neurologists (preferably ALS specialists)
- Respiratory therapists
- Physical therapists
- Occupational therapists
- Speech-language pathologists
- Nutritionists
- Social workers
- Palliative care specialists
- Mental health professionals
Disease-Modifying Medications:
- Riluzole (Rilutek): First FDA-approved drug (1995)
- Mechanism: Glutamate release inhibition
- Efficacy: Extends survival by approximately 2-3 months
- Dosage: 50mg twice daily
- Edaravone (Radicava): FDA approved in 2017
- Mechanism: Free radical scavenger/antioxidant
- Efficacy: Slows functional decline by approximately 33%
- Administration: Intravenous infusion cycles
- Sodium phenylbutyrate/taurursodiol (Relyvrio): FDA approved in 2022
- Mechanism: Reduces endoplasmic reticulum stress and mitochondrial dysfunction
- Efficacy: Preliminary data suggests slowing of functional decline
- Administration: Oral medication
Symptomatic Management:
- Spasticity: Baclofen, tizanidine, benzodiazepines
- Cramps: Quinine, mexiletine, magnesium supplements
- Excessive saliva: Anticholinergics, botulinum toxin, radiation therapy
- Pseudobulbar affect: Dextromethorphan/quinidine (Nuedexta)
- Pain: Standard analgesics, gabapentin for neuropathic components
- Depression/anxiety: SSRIs, SNRIs, counseling
- Fatigue: Modafinil, amantadine, energy conservation techniques
- Insomnia: Melatonin, trazodone, careful use of sedatives
Medications, Surgeries, and Therapies
Respiratory Management:
- Non-invasive ventilation (NIV): Typically BiPAP
- Invasive ventilation (tracheostomy) when NIV insufficient
- Mechanical insufflation-exsufflation (cough assist devices)
- Supplemental oxygen (used cautiously)
- Respiratory muscle training in early stages
Nutritional Management:
- Dietary modifications for swallowing difficulties
- High-calorie, high-protein diets
- Percutaneous endoscopic gastrostomy (PEG) tube placement
- Radiologically inserted gastrostomy (RIG) for respiratory-compromised patients
- Nutritional supplements
Rehabilitative Therapies:
- Physical therapy: Stretching, range of motion, assistive device training
- Occupational therapy: Adaptive equipment, home modifications
- Speech therapy: Communication strategies, augmentative devices
- Swallowing therapy: Techniques to reduce aspiration risk
Assistive Technology:
- Mobility devices: Canes, walkers, wheelchairs, mobility scooters
- Communication devices: Voice amplifiers, text-to-speech, eye-gaze technology
- Environmental controls: Voice or switch-activated systems
- Adaptive eating utensils and personal care equipment
Palliative Care:
- Symptom management
- Advanced care planning
- Emotional and spiritual support
- Family support and education
- Hospice coordination when appropriate
Emerging Treatments and Clinical Trials
Gene Therapy Approaches:
- Antisense oligonucleotides (ASOs) for SOD1 and C9orf72 mutations
- CRISPR/Cas9 gene editing techniques
- AAV-delivered gene therapies
Stem Cell Therapies:
- Mesenchymal stem cell transplantation
- Neural progenitor cell implantation
- Induced pluripotent stem cell (iPSC) derived motor neurons
Novel Pharmacological Approaches:
- Targeting neuroinflammation (NP001, masitinib)
- Autophagy enhancers (arimoclomol)
- Protein aggregation inhibitors
- Mitochondrial function enhancers
- Neurotrophic factors (BDNF, GDNF delivery methods)
Immunotherapy Approaches:
- Regulatory T-cell therapy (Tregs)
- Microglial modulation
- Monoclonal antibody therapies
Technology-Based Interventions:
- Brain-computer interfaces for communication and control
- Exoskeletons for mobility assistance
- Virtual reality for cognitive maintenance
- Robotic feeding and care assistance
Alternative and Complementary Approaches Under Study:
- Medical cannabis derivatives
- Ketogenic diet
- Intermittent fasting protocols
- Exercise protocols (carefully designed)
- Acupuncture
- Traditional Chinese medicine compounds
As of 2023-2024, there were over 100 active clinical trials for ALS treatments, indicating the significant research focus on this devastating disease.
9. Prevention & Precautionary Measures
Preventing ALS
ALS is not currently preventable, as most cases occur sporadically without clearly identifiable causes. However, some approaches may theoretically reduce risk or delay onset:
Current Limitations:
- No proven prevention strategies exist
- Most risk factors are non-modifiable (age, genetics)
- Multifactorial nature makes prevention challenging
Theoretical Genetic Prevention:
- Genetic counseling for affected families
- Preimplantation genetic diagnosis for known mutations
- Emerging gene therapy approaches for presymptomatic carriers (experimental)
Lifestyle Changes and Environmental Precautions
While no lifestyle modifications have been proven to prevent ALS, some may potentially reduce risk based on epidemiological evidence:
Dietary Considerations:
- Mediterranean diet (anti-inflammatory)
- Adequate vitamin E intake
- Omega-3 fatty acid consumption
- Vitamin D sufficiency
- Antioxidant-rich foods
Physical Activity:
- Moderate physical activity (extreme exercise is controversial)
- Avoiding repetitive head/neck trauma (especially in contact sports)
- Proper body mechanics to avoid physical strain
Environmental Precautions:
- Avoiding exposure to:
- Certain pesticides and herbicides
- Lead and other heavy metals
- Excessive electromagnetic fields (controversial)
- Cyanobacterial toxins in water bodies
Other Potentially Protective Factors:
- Maintaining healthy weight
- Smoking cessation
- Stress management
- Adequate sleep
- Blood pressure control
Vaccines or Preventive Screenings
Current Status:
- No vaccines exist for ALS prevention
- No recommended screening protocols for general population
- No validated biomarkers for risk assessment
Potential Future Approaches:
- Genetic screening for high-risk families
- Blood-based biomarker panels under development
- Specialized imaging for those with family history
- Monitoring of high-risk professions (athletes, military)
Research Directions:
- Developing predictive algorithms combining:
- Genetic markers
- Environmental exposure history
- Biological markers (NfL, microRNAs)
- Imaging findings
- Clinical assessment
Prevention research faces significant challenges due to the relatively low incidence of ALS, its heterogeneous nature, and its typically sporadic occurrence. Current focus remains on early detection and intervention rather than true prevention.
10. Global & Regional Statistics
Incidence and Prevalence Rates Globally
Global Average Incidence:
- 1-2.6 new cases per 100,000 people annually
Global Average Prevalence:
- 4.1-8.4 per 100,000 people (global average)
Regional Variations in Incidence (per 100,000):
- North America: 1.5-2.0
- Europe: 2.0-2.4
- East Asia: 0.7-0.8
- South Asia: 0.3-0.5
- Africa: 0.5-0.8 (limited data available)
- Australia/New Zealand: 1.9-2.3
- South America: 0.7-1.5
Notable Outliers:
- Western Pacific focus: Historical cluster in Guam (50× higher rates)
- Kii Peninsula, Japan: 3-4× higher than Japanese average
- Parts of Sardinia: 2-3× higher than Italian average
Mortality and Survival Rates
Global Mortality Patterns:
- ALS is universally fatal without ventilation support
- Annual mortality rate: Approximately 1.0-1.8 per 100,000 population
Survival Rates:
- Median survival from diagnosis: 2-4 years globally
- 5-year survival rate: 20-30%
- 10-year survival rate: 5-10%
- 20-year survival rate: 1-3%
Factors Affecting Survival:
- Age at onset (younger = longer survival)
- Site of onset (limb > bulbar > respiratory)
- Respiratory status at diagnosis
- Rate of functional decline in first 3 months
- Cognitive impairment (shorter survival if present)
- Access to multidisciplinary care
- Genetic subtype (C9orf72 typically worse prognosis than sporadic)
- Nutritional status
- Country’s healthcare system and access to care
Country-wise Comparison and Trends
Highest ALS Rates:
- Finland
- Sweden
- Norway
- United States
- United Kingdom
- France
- Netherlands
Lowest Reported Rates:
- Mexico
- China
- Iran
- South Asian countries
- Most African countries (though underdiagnosis is likely)
Temporal Trends:
- Global incidence increasing approximately 1-2% per decade
- Most pronounced increase in:
- East Asia (possibly due to improved diagnosis)
- African countries (as healthcare improves)
- Aging populations globally
Healthcare System Impact:
- Survival longest in countries with:
- Universal healthcare access
- Specialized ALS centers
- Early access to NIV and nutritional support
- Homecare support systems
Research Investment by Region:
- North America: Highest per capita research funding
- European Union: Strong coordinated research networks
- Japan: Significant biomarker and genetic research
- Australia: Strong clinical trial infrastructure
- China: Rapidly growing research output
The global landscape of ALS shows some geographic patterns that suggest both genetic and environmental contributors, though improved diagnosis in developing regions may be reducing apparent disparities.
11. Recent Research & Future Prospects
Latest Advancements in Treatment and Research
Genetic Understanding:
- Expanded gene panels identifying new causative and risk-modifying genes
- Polygenic risk scores development
- Single-cell transcriptomics revealing cell-specific vulnerabilities
- Epigenetic modifications in ALS pathogenesis
Biomarker Development:
- Neurofilament light chain (NfL) validation for diagnosis and monitoring
- MicroRNA panels showing diagnostic potential
- Skin biopsy methods for TDP-43 pathology detection
- Imaging biomarkers for upper motor neuron assessment
- Digital biomarkers using smartphone and wearable data
Pathophysiology Insights:
- Expanded understanding of:
- RNA processing abnormalities
- Nuclear transport defects
- Liquid-liquid phase separation in protein aggregation
- Prion-like spreading of pathological proteins
- Microglial and astrocyte contributions
- Gut microbiome influences
Treatment Innovations:
- Antisense oligonucleotides for genetic forms:
- Tofersen for SOD1 ALS (demonstrated SOD1 protein reduction)
- BIIB078 for C9orf72 ALS (in clinical trials)
- Oral version of edaravone improving accessibility
- Novel delivery systems crossing blood-brain barrier
- Repurposed medications showing promise:
- Sodium phenylbutyrate combinations
- Metformin in specific genetic subtypes
- Ursodeoxycholic acid derivatives
Clinical Care Advances:
- Telemedicine protocols for ALS care
- Multidisciplinary clinic models showing survival benefits
- Respiratory monitoring using home-based systems
- Advanced communication devices with improved algorithms
- Palliative care integration from diagnosis
Ongoing Studies and Future Medical Possibilities
Major Clinical Trials:
- Platform trials testing multiple drugs simultaneously
- Biomarker-guided treatment selection trials
- Combination therapy approaches
- Different treatment strategies based on ALS subtype
Technological Developments:
- Advanced brain-computer interfaces
- Exoskeleton technology for mobility
- Voice banking and digital voice preservation
- Smart home technology for independence
- Assistive robots for care support
Experimental Approaches:
- Parabiosis studies (young-old blood exchange)
- Gut microbiome modification
- Metabolic reprogramming
- Mitochondrial transplantation
- Ultrasound-guided blood-brain barrier opening for drug delivery
Scientific Infrastructure:
- ALS biobanks and collaborative networks
- Harmonized data collection across centers
- Patient-reported outcome measures
- Virtual clinical trials
- AI for digital biosignature identification
Potential Cures or Innovative Therapies Under Development
Gene Therapy Approaches:
- AAV9-mediated gene replacement for genetic forms
- CRISPR/Cas9 gene editing for mutation correction
- RNA-targeting therapies beyond ASOs
- Suppression of C9orf72 repeat expansions
Stem Cell Therapies:
- Neural progenitor cells secreting growth factors
- Direct motor neuron replacement (challenging but advancing)
- Engineered mesenchymal stem cells with enhanced properties
- iPSC-derived astrocytes to support motor neurons
Immunotherapeutic Approaches:
- Expanded Treg cell therapy
- Microglial reprogramming
- Custom-designed antibodies targeting misfolded proteins
- Vaccination approaches against toxic protein species
Regenerative Medicine:
- Growth factor delivery systems
- Small molecules promoting axonal regeneration
- Scaffolds for neural regrowth
- Electrical stimulation to maintain neuromuscular connections
Combination Approaches:
- Multi-drug regimens targeting different pathways
- Gene therapy plus small molecule combinations
- Immunomodulation with neuroprotective agents
- Symptomatic plus disease-modifying strategies
The future of ALS treatment is likely to involve personalized approaches based on genetic profiles, biomarkers, and disease subtypes, with combination therapies addressing multiple aspects of this complex disorder.
12. Interesting Facts & Lesser-Known Insights
Uncommon Knowledge About ALS
Historical Perspectives:
- Lou Gehrig may not have had the disease named after him: Recent research suggests he might have suffered from chronic traumatic encephalopathy (CTE) from baseball injuries rather than ALS
- Famous physicist Stephen Hawking lived with ALS for 55 years, an extraordinarily rare case
- The first description of ALS by Charcot included detailed drawings that remain accurate today
- ALS was once called “Charcot disease” in many European countries
Biological Peculiarities:
- Eye movements are preserved even in advanced ALS, allowing for eye-tracking communication
- Bladder and bowel control typically remain intact until very late stages
- The “split hand syndrome” in ALS shows peculiar vulnerability of thumb muscles
- Heart muscle is almost never affected, even when skeletal muscles are severely atrophied
- Sensory neurons are typically spared, leading to the tragic situation of fully feeling a body that cannot move
Geographic and Demographic Patterns:
- Professional athletes have 4-6× higher risk in some sports
- Military veterans have approximately double the risk of the general population
- Men get ALS more often than women until age 65, after which the rates equalize
- Lifetime risk of ALS is approximately 1 in 300 for men and 1 in 400 for women
- Incidence peaks at two different age ranges: 40s and 70s, suggesting possibly different disease mechanisms
Research Challenges:
- Over 50 clinical trials have failed despite promising preclinical results
- Animal models of ALS are limited in predicting human responses
- The disease can present differently in almost every patient
- At least 5 major theoretical mechanisms compete to explain ALS pathogenesis
Myths and Misconceptions vs. Medical Facts
Myth: ALS always progresses rapidly and leads to death within 2-3 years. Fact: About 10-20% of patients have slow progression, with survival beyond 10 years.
Myth: ALS affects the mind and causes dementia in most patients. Fact: Only about 15-20% of ALS patients develop frontotemporal dementia, though subtle cognitive changes may be more common.
Myth: ALS is strictly a disease of motor neurons. Fact: Research shows involvement of other cell types including astrocytes, microglia, and oligodendrocytes in disease pathogenesis.
Myth: ALS is purely a genetic disease passed down in families. Fact: Only 5-10% of cases are clearly familial, though genetic contributions exist in sporadic cases.
Myth: There are no treatments available for ALS. Fact: While there is no cure, FDA-approved treatments can slow progression, and symptomatic management significantly improves quality of life.
Myth: ALS diagnosis means immediate loss of independence. Fact: The disease typically progresses in stages, with many patients maintaining significant independence for months or years.
Myth: ALS affects everyone in the same way. Fact: The disease has remarkable variability in onset site, progression rate, and symptom profile.
Myth: ALS research has made little progress. Fact: Significant advances in understanding genetics, pathophysiology, and potential therapeutic targets have occurred, especially in the past decade.
Impact on Specific Populations or Professions
Professional Athletes:
- Football players show 4× increased risk in multiple studies
- Italian soccer players demonstrated 6× higher rates
- Professional athletes often experience earlier onset
- Hypothesis links repetitive head/neck trauma to risk
Military Personnel:
- Gulf War veterans show 2× higher risk
- Military personnel exposed to certain pesticides, lead, and chemicals show increased rates
- Some studies suggest physical exertion under stress may be a trigger
- Military ALS often affects younger individuals
Performing Artists:
- Notable cases in musicians, actors, and dancers
- Hypothesis that intense, repetitive motor activity might contribute
- Some famous cases: Jason Becker (guitarist), Sesame Street creator Jon Stone, actor David Niven
Scientific Community Response:
- The Ice Bucket Challenge raised over $115 million for ALS research
- Created unprecedented research momentum
- Led to discovery of 5 new ALS-linked genes
- Established the largest ALS sequencing database
- Funded development of new animal models
Impact on Caregivers:
- ALS caregiving ranks among the most demanding of all conditions
- Average of 11-15 daily hours of care in advanced stages
- Significant physical, emotional, and financial burden
- Caregivers show high rates of depression (30-50%)
- Specialized programs developing to support ALS caregivers
The impact of ALS extends far beyond the individual, affecting families, healthcare systems, and specific communities, while continuing to catalyze remarkable scientific collaboration in the search for effective treatments and ultimately a cure.