⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
Comprehensive Report on Amyotrophic Lateral Sclerosis (ALS)
1. Overview
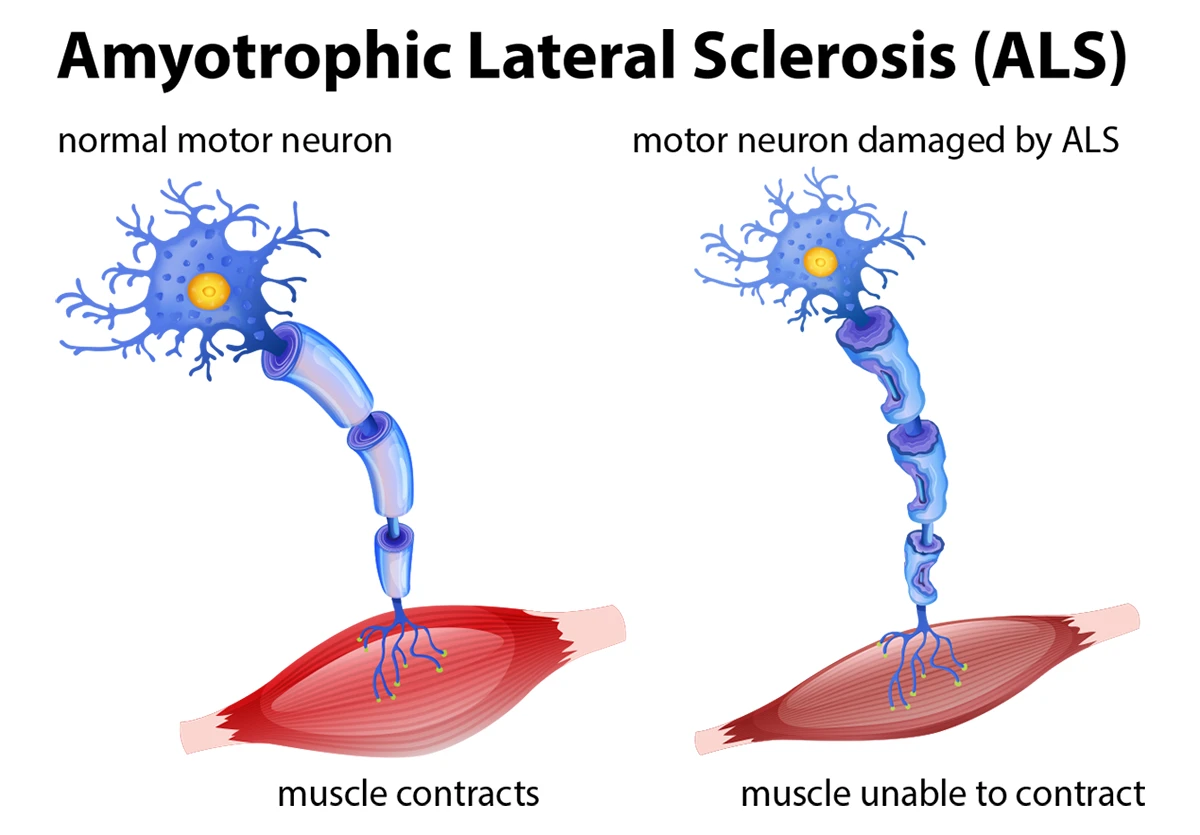
Amyotrophic Lateral Sclerosis (ALS), also known as Lou Gehrig’s disease or Motor Neuron Disease in some countries, is a progressive neurodegenerative disorder characterized by the gradual degeneration and death of motor neurons. Motor neurons are nerve cells that extend from the brain to the spinal cord and to muscles throughout the body, controlling voluntary muscle movement.
ALS primarily affects two types of motor neurons:
- Upper motor neurons: Located in the brain’s motor cortex
- Lower motor neurons: Located in the brainstem and spinal cord
As these motor neurons degenerate and die, they stop sending messages to the muscles, which leads to muscle weakening, twitching (fasciculations), and an inability to initiate and control voluntary movements. Eventually, this results in paralysis.
The disease primarily affects the motor system, but increasingly research shows it can affect cognitive function in some patients. The sensory neurons, which process sensory information such as touch, pain, temperature, and position, usually remain intact. Similarly, the neurons controlling eye movements and bladder and bowel functions are typically spared.
Prevalence and significance:
- In the United States, approximately 5,000 people are diagnosed with ALS each year
- The prevalence is about 5 cases per 100,000 people in the general population
- ALS is considered a rare disease, affecting approximately 30,000 Americans at any given time
- It is the most common motor neuron disease in adults
- The disease has profound significance due to its devastating progression, high mortality rate, and substantial impact on quality of life for patients and caregivers
2. History & Discoveries
Initial Identification
ALS was first described in 1869 by the renowned French neurologist Jean-Martin Charcot. Through careful clinical observation and post-mortem studies, Charcot identified and documented the relationship between the symptoms of the disease and underlying neurological damage.
The name “Amyotrophic Lateral Sclerosis” reflects Charcot’s observations:
- “Amyotrophic” refers to the muscle atrophy that occurs
- “Lateral” refers to the areas of the spinal cord affected by the disease
- “Sclerosis” refers to the scarring or hardening of the spinal cord’s lateral columns
Public Awareness
The disease gained significant public attention in the United States when baseball legend Lou Gehrig was diagnosed with ALS in 1939. His famous “luckiest man on the face of the earth” speech at Yankee Stadium and subsequent death from the disease in 1941 put ALS in the public consciousness, leading to the alternative name “Lou Gehrig’s disease” in North America.
Major Breakthroughs
- 1940s-1950s: Basic characterization of the disease’s pathology and progression
- 1962: Development of the first ALS clinical rating scale by Norris
- 1993: Discovery of mutations in the superoxide dismutase 1 (SOD1) gene as the first genetic cause of familial ALS
- 1995: FDA approval of riluzole (Rilutek), the first drug shown to extend survival in ALS patients
- 2006: Discovery of TDP-43 protein aggregates in ALS patients
- 2011: Identification of C9orf72 repeat expansions as the most common genetic cause of both familial ALS and frontotemporal dementia
- 2014: The ALS Ice Bucket Challenge went viral, raising over $115 million for ALS research
- 2017: FDA approval of edaravone (Radicava), the second drug for ALS treatment
- 2022: FDA approval of AMX0035 (Relyvrio), a combination of sodium phenylbutyrate and taurursodiol
Evolution of Understanding
Our understanding of ALS has evolved significantly:
- Initially viewed as a pure motor neuron disease
- Now recognized as a spectrum disorder with varying degrees of motor and cognitive involvement
- Evolved from being considered a single disease to being understood as a heterogeneous condition with multiple subtypes
- Shifted from focusing solely on motor neurons to recognizing it involves multiple cellular and molecular pathways
- Advanced from having no treatments to having three FDA-approved medications with many more in clinical trials
3. Symptoms
Early Symptoms
The initial symptoms of ALS can be subtle and often vary depending on which motor neurons are affected first. Early symptoms typically include:
Limb-onset ALS (approximately 70% of cases):
- Muscle weakness in hands, arms, or legs
- Muscle twitching (fasciculations)
- Muscle cramps
- Stiffness or rigidity in muscles
- Difficulty with fine motor tasks like buttoning shirts or turning keys
- Tripping, stumbling, or foot drop
- Decreased coordination
Bulbar-onset ALS (approximately 25% of cases):
- Slurred speech (dysarthria)
- Difficulty swallowing (dysphagia)
- Excessive saliva due to difficulty swallowing
- Voice changes or hoarseness
- Inappropriate emotional responses (pseudobulbar affect)
Truncal/respiratory-onset ALS (approximately 5% of cases):
- Shortness of breath
- Difficulty with trunk mobility
- Impaired breathing, especially when lying down
Advanced-Stage Symptoms
As the disease progresses, symptoms become more severe and widespread:
- Widespread muscle weakness leading to paralysis
- Severe speech impairment or inability to speak
- Significant difficulty swallowing, requiring feeding tube placement
- Respiratory insufficiency requiring ventilatory support
- Weight loss and muscle atrophy
- Inability to perform activities of daily living
- In some cases, cognitive changes ranging from mild impairment to frontotemporal dementia
Symptom Progression
ALS typically progresses in the following pattern:
- Initial Stage: Focal weakness in one region (limb or bulbar)
- Middle Stage: Spread to adjacent regions and increasing disability
- Late Stage: Generalized weakness with respiratory involvement
- Terminal Stage: Complete paralysis and respiratory failure
The rate of progression varies significantly between individuals. On average, survival from symptom onset ranges from 3-5 years, but approximately 10% of patients live 10 years or longer. Notably, physicist Stephen Hawking lived with ALS for 55 years, representing an exceptional case.
Factors that may indicate a slower disease progression include:
- Younger age at onset
- Limb onset (versus bulbar onset)
- Primarily upper motor neuron involvement
- Absence of cognitive impairment
- Certain genetic variants
4. Causes
The exact cause of ALS remains incompletely understood, but research has identified several biological mechanisms and risk factors that contribute to the disease.
Biological Mechanisms
Several cellular and molecular mechanisms appear to contribute to motor neuron degeneration in ALS:
- Protein misfolding and aggregation: Abnormal protein accumulation, particularly TDP-43, FUS, and SOD1 proteins
- Glutamate excitotoxicity: Excessive stimulation of motor neurons by the neurotransmitter glutamate
- Oxidative stress: Damage to cells from free radicals and reactive oxygen species
- Mitochondrial dysfunction: Impaired cellular energy production
- Disrupted axonal transport: Problems with the movement of cellular components along motor neuron axons
- Neuroinflammation: Activation of inflammatory cells in the nervous system
- RNA processing defects: Abnormalities in how genetic information is processed
- Cytoskeletal abnormalities: Disruption of the internal structure of motor neurons
Genetic Factors
Approximately 5-10% of ALS cases are familial (inherited), while 90-95% are sporadic (occurring without a family history). However, genetic factors may contribute to both types:
Major genes implicated in ALS:
- C9orf72: Hexanucleotide repeat expansion – most common genetic cause (30-40% of familial ALS, 5-10% of sporadic ALS)
- SOD1: First discovered ALS gene (12-20% of familial ALS, 1-2% of sporadic ALS)
- TARDBP (TDP-43): Important in RNA processing (4-5% of familial ALS)
- FUS: Similar function to TDP-43 (4-5% of familial ALS)
- NEK1: Recently identified through genome-wide studies (3% of all ALS cases)
- Other genes: TBK1, OPTN, VCP, UBQLN2, PFN1, TUBA4A, ANXA11, and many others
The diversity of implicated genes suggests multiple pathways can lead to motor neuron degeneration.
Environmental Factors
Several environmental exposures have been studied as potential contributors to ALS risk:
- Heavy metal exposure: Lead, mercury, and other metals
- Pesticides and agricultural chemicals
- Military service: Particularly Gulf War veterans
- Head trauma and intense physical exertion: Particularly relevant for the potential link to professional athletes
- Electromagnetic field exposure: Still controversial
- Cyanobacterial toxins: Particularly in the Western Pacific ALS clusters
- Viral infections: Some evidence for human endogenous retroviruses
Most likely, ALS develops through a complex interaction between genetic susceptibility and environmental triggers in most cases.
5. Risk Factors
Several factors appear to increase the risk of developing ALS:
Demographic Factors
- Age: Most cases occur between ages 55-75, with median age of onset around 64-67 years
- Gender: Men have a slightly higher risk than women (ratio approximately 1.5:1), though this difference diminishes with age
- Race/Ethnicity: Generally higher rates in Caucasians than in African, Asian, or Hispanic populations
Genetic Factors
- Family history: Having a first-degree relative with ALS increases risk
- Certain genetic variants: Even in sporadic cases, risk alleles may contribute
- Genetic background: May interact with environmental exposures
Lifestyle and Environmental Factors
- Smoking: Associated with approximately 1.5-fold increased risk
- Military service: Veterans have approximately 1.5-2 times higher risk than civilians
- Occupational exposures:
- Agricultural work and pesticide exposure
- Certain industrial chemicals
- Lead and other heavy metals
- Electric shock injury
- Physical activity: Controversial, with conflicting evidence on whether intense exercise is protective or harmful
- Head trauma: Multiple concussions may increase risk
- Obesity and metabolic factors: Lower body mass index (BMI) before diagnosis associated with higher risk
Pre-existing Conditions
- Frontotemporal dementia: Genetic and clinical overlap with ALS
- Autoimmune diseases: Some evidence for shared mechanisms
- Prior polio infection: Suggested but not conclusively proven
- Metabolic disorders: Diabetes may affect progression, though the relationship with risk is unclear
The interplay between these factors is complex, and most patients with ALS have multiple risk factors that combine to trigger the disease.
6. Complications
ALS leads to numerous complications as the disease progresses:
Respiratory Complications
- Respiratory insufficiency: Progressive weakening of the diaphragm and intercostal muscles
- Respiratory failure: The most common cause of death in ALS
- Aspiration pneumonia: Due to impaired swallowing and weakened cough
- Sleep-disordered breathing: Including sleep apnea and hypoventilation
- Increased susceptibility to respiratory infections
Nutritional Complications
- Dysphagia (difficulty swallowing): Leading to inadequate food and fluid intake
- Malnutrition and weight loss: Accelerated by increased metabolic demands
- Dehydration: Due to inability to swallow liquids safely
- Choking risk: Impaired swallowing mechanism increases aspiration risk
Mobility and Musculoskeletal Complications
- Progressive paralysis: Eventually affecting most voluntary muscles
- Contractures: Permanent tightening of muscles, tendons, and joints
- Pressure sores: Due to immobility and inability to change positions
- Deep vein thrombosis: Blood clots from immobility
- Muscle atrophy and spasticity: Causing pain and discomfort
Communication Complications
- Anarthria: Complete loss of speech
- Social isolation: Due to communication difficulties
- Emotional impact: Frustration from inability to communicate
Psychological and Cognitive Complications
- Depression and anxiety: Common responses to diagnosis and progression
- Pseudobulbar affect: Uncontrollable laughing or crying unrelated to emotional state
- Cognitive changes: From mild executive dysfunction to frontotemporal dementia
- Existential distress: Confronting mortality and loss of function
Other Physical Complications
- Autonomic dysfunction: Problems with blood pressure regulation, temperature control
- Excessive saliva or dry mouth: Due to bulbar muscle weakness
- Urinary urgency: Though incontinence is uncommon until late stages
- Sleep disturbances: Due to physical discomfort and breathing difficulties
Impact on Life Expectancy
- Median survival is 3-5 years from diagnosis
- Approximately 20% of patients live 5 years
- About 10% survive 10 years or more
- Survival predictors include:
- Age at onset (younger = better prognosis)
- Site of onset (limb onset generally better than bulbar)
- Rate of disease progression
- Respiratory function
- Presence of genetic mutations
- Access to multidisciplinary care
7. Diagnosis & Testing
Diagnosing ALS remains challenging due to the lack of a definitive test. The process typically involves:
Diagnostic Approach
- Clinical evaluation: Thorough neurological examination looking for upper and lower motor neuron signs
- Detailed medical history: Including symptom progression, family history, and exposure history
- Application of diagnostic criteria: Most commonly the revised El Escorial criteria or the Awaji criteria
- Exclusion of ALS mimics: Conditions that present with similar symptoms
Laboratory Tests
- Blood and urine tests: To rule out metabolic, infectious, or autoimmune disorders
- Cerebrospinal fluid analysis: May help exclude inflammatory conditions
- Genetic testing: Particularly for patients with family history or early-onset disease
- Biomarker testing: Emerging tests measuring neurofilament levels and other biomarkers
Electrophysiological Studies
- Electromyography (EMG): Measures electrical activity in muscles; crucial for diagnosis
- Nerve conduction studies (NCS): Evaluates peripheral nerve function
- Transcranial magnetic stimulation: Assesses upper motor neuron function
- Motor unit number estimation (MUNE): Quantifies surviving motor neurons
Imaging Studies
- Magnetic Resonance Imaging (MRI): Primarily to rule out other conditions like spinal cord compression
- Advanced neuroimaging: Includes diffusion tensor imaging and functional MRI
- Muscle ultrasound: Can detect fasciculations and assess muscle architecture
- PET scanning: Sometimes used in research settings
Other Diagnostic Procedures
- Muscle biopsy: Occasionally performed to rule out muscle diseases
- Neuropsychological testing: To assess cognitive function
- Respiratory function tests: Including forced vital capacity (FVC) and sniff nasal inspiratory pressure
- Swallowing studies: To assess bulbar function
Diagnostic Challenges
- Diagnostic delay: Average of 12-14 months from symptom onset to diagnosis
- Early stages: Symptoms may be subtle and localized
- ALS variants: Some presentations differ from classic ALS
- Mimicking conditions: Many disorders can initially resemble ALS:
- Multifocal motor neuropathy
- Cervical myelopathy
- Kennedy’s disease (spinobulbar muscular atrophy)
- Post-polio syndrome
- Myasthenia gravis
- Inclusion body myositis
- Primary lateral sclerosis
- Progressive muscular atrophy
Diagnostic Certainty
The El Escorial criteria classify diagnostic certainty into categories:
- Definite ALS: Upper and lower motor neuron signs in three regions
- Probable ALS: Upper and lower motor neuron signs in two regions
- Possible ALS: Upper and lower motor neuron signs in one region
- Suspected ALS: Lower motor neuron signs only in two or more regions
Early diagnosis is crucial for timely intervention but must be balanced against the profound impact of an ALS diagnosis.
8. Treatment Options
While there is currently no cure for ALS, treatment approaches focus on slowing disease progression, managing symptoms, and improving quality of life through a multidisciplinary approach.
FDA-Approved Medications
Riluzole (Rilutek):
- Approved in 1995
- Mechanism: Reduces glutamate excitotoxicity
- Benefits: Extends survival by approximately 2-3 months on average
- Administration: Oral tablet, 50mg twice daily
- Side effects: Nausea, dizziness, liver enzyme elevations
Edaravone (Radicava):
- Approved in 2017
- Mechanism: Antioxidant, reduces oxidative stress
- Benefits: Slows functional decline by about 33% in a subset of patients
- Administration: Initially intravenous infusion, now also available as oral formulation
- Side effects: Bruising, gait disturbances, headache
Sodium phenylbutyrate-taurursodiol (Relyvrio/AMX0035):
- Approved in 2022
- Mechanism: Targets endoplasmic reticulum and mitochondrial stress
- Benefits: Slows functional decline and extends survival
- Administration: Oral suspension
- Side effects: Diarrhea, abdominal pain, nausea
Symptom Management
Muscle cramps and spasticity:
- Baclofen, tizanidine, or benzodiazepines
- Physical therapy and stretching
Excessive saliva:
- Anticholinergic medications (glycopyrrolate, atropine)
- Botulinum toxin injections to salivary glands
- Suction devices
Pain:
- Non-steroidal anti-inflammatory drugs
- Neuropathic pain medications (gabapentin, pregabalin)
- Opioids for severe pain
Pseudobulbar affect:
- Dextromethorphan/quinidine (Nuedexta)
- Antidepressants
Depression and anxiety:
- Selective serotonin reuptake inhibitors (SSRIs)
- Psychotherapy
- Support groups
Sleep disturbances:
- Treatment of underlying respiratory issues
- Sleep medications when appropriate
Supportive Therapies
Physical therapy:
- Range of motion exercises
- Strengthening of unaffected muscles
- Gait training and fall prevention
Occupational therapy:
- Adaptive equipment recommendations
- Home modification guidance
- Energy conservation techniques
Speech therapy:
- Communication strategies
- Voice banking
- Alternative augmentative communication devices
Respiratory therapy:
- Breathing exercises
- Airway clearance techniques
- Non-invasive ventilation (NIV) guidance
Nutritional support:
- Dietary modifications
- Nutritional supplements
- Consultation regarding feeding tube placement
Assistive Technology
Mobility devices:
- Ankle-foot orthoses
- Walkers and wheelchairs
- Transfer equipment
Communication devices:
- Speech generating devices
- Eye-tracking technology
- Brain-computer interfaces (emerging)
Respiratory support:
- Non-invasive ventilation (BiPAP)
- Mechanical insufflation-exsufflation (cough assist)
- Invasive ventilation (tracheostomy) in selected cases
Home adaptations:
- Smart home technology
- Environmental control systems
- Adaptive utensils and tools
Emerging Treatments and Clinical Trials
Gene therapy approaches:
- Antisense oligonucleotides for genetic forms
- Gene silencing techniques
- CRISPR gene editing (preclinical)
Stem cell therapy:
- Mesenchymal stem cells
- Neural progenitor cells
- Induced pluripotent stem cells
Targeting neuroinflammation:
- NP001, Masitinib
- Microglial modulators
Neuroprotective agents:
- Multiple compounds in development targeting various pathways
Compounds targeting protein aggregation:
- Addressing TDP-43 and SOD1 aggregates
Multidisciplinary Care
Research shows that multidisciplinary ALS clinics improve quality of life and may extend survival. These clinics typically include:
- Neurologists specializing in ALS
- Respiratory therapists
- Physical and occupational therapists
- Speech-language pathologists
- Nutritionists
- Social workers
- Palliative care specialists
- Psychologists
- Assistive technology experts
End-of-Life Care
- Advance care planning: Discussing preferences for end-of-life interventions
- Palliative care: Focusing on comfort and quality of life
- Hospice care: When prognosis is six months or less
- Psychological and spiritual support: For both patients and families
9. Prevention & Precautionary Measures
Since the exact causes of ALS remain unclear, there are currently no proven methods to prevent the disease. However, researchers have identified several approaches that may potentially reduce risk or delay onset:
Potential Risk Reduction Strategies
Nutritional factors:
- Mediterranean diet rich in antioxidants
- Adequate vitamin E intake
- Omega-3 fatty acids
- Vitamin D maintenance
- Proper hydration
Physical activity:
- Moderate, regular exercise appears beneficial
- Avoiding excessive physical exertion or trauma
- Maintaining overall physical fitness without overtraining
Avoiding environmental toxins:
- Limiting exposure to pesticides and herbicides
- Reducing contact with industrial chemicals and solvents
- Proper handling of potential neurotoxins in occupational settings
- Water filtration in areas with known environmental concerns
Smoking cessation:
- Clear evidence links smoking to increased ALS risk
Head protection:
- Preventing head trauma in sports and occupational settings
- Proper concussion management
For Individuals with Genetic Risk
Genetic counseling:
- Understanding inheritance patterns and risks
- Family planning considerations
- Psychological support for at-risk individuals
Genetic testing considerations:
- Testing options for known ALS genes
- Implications for family members
- Ethical considerations and privacy concerns
Experimental interventions:
- Clinical trials specifically for presymptomatic gene carriers
- Emerging preventive gene therapies
General Neurological Health
Cardiovascular health:
- Managing blood pressure, cholesterol, and glucose levels
- Regular cardiovascular exercise
Brain health practices:
- Cognitive engagement and mental stimulation
- Adequate sleep
- Stress management techniques
Anti-inflammatory lifestyle:
- Reducing chronic inflammation through diet and exercise
- Avoiding obesity and metabolic syndrome
Limitations in Prevention
It’s important to note several key limitations:
- No preventive measures have been definitively proven to reduce ALS risk
- ALS likely results from complex interactions between multiple factors
- Individual genetic susceptibility plays a significant role
- No vaccines or prophylactic medications currently exist
- Screening programs for the general population are not recommended
Future Directions in Prevention
Emerging research areas that may lead to preventive strategies include:
- Biomarker identification for presymptomatic detection
- Personalized risk assessment based on genetic profiling
- Targeted interventions for high-risk individuals
- Environmental monitoring in areas with elevated ALS incidence
- Occupation-specific safety measures for at-risk professions
10. Global & Regional Statistics
ALS epidemiology varies across different regions and populations:
Global Incidence and Prevalence
- Worldwide incidence: Approximately 1-2.6 cases per 100,000 person-years
- Global prevalence: 4-6 cases per 100,000 population
- Annual new cases worldwide: Estimated at 140,000-150,000
- Geographic distribution: Generally higher in Western countries, lower in East Asia, South Asia, and Africa
Regional Variations
North America
United States:
- Incidence: 1.5-2.5 per 100,000 person-years
- Prevalence: 5.0-5.5 per 100,000 population
- Approximately 5,000-6,000 new diagnoses annually
- Estimated 30,000 Americans living with ALS
Canada:
- Similar rates to the United States
- Slightly higher rates in certain provinces
Europe
Overall European rates:
- Incidence: 2.1-3.0 per 100,000 person-years
- Prevalence: 5.4-7.3 per 100,000 population
Regional variations:
- Higher rates in Scandinavian countries
- North-to-south gradient with lower rates in Southern Europe
Asia
Japan:
- Incidence: 1.2-1.5 per 100,000 person-years
- Higher rates in the Kii Peninsula (historical cluster)
China:
- Incidence: 0.6-0.8 per 100,000 person-years
- Lower than Western countries
South Asia:
- Generally lower reported rates, potentially due to underdiagnosis
- Incidence: 0.2-0.5 per 100,000 person-years in India
Other Regions
Australia/New Zealand:
- Rates similar to Europe and North America
South America:
- Limited epidemiological data
- Rates appear slightly lower than North America
Africa:
- Sparse data available
- Likely underreported due to healthcare access limitations
ALS Clusters and High-Incidence Regions
Western Pacific ALS clusters:
- Guam (Chamorro people): Historically very high rates, now declining
- Kii Peninsula, Japan: 3-4 times the national average
- New Guinea: Elevated rates in certain indigenous populations
Suspected environmental clusters:
- Gulf War veterans: 1.5-2 times higher risk
- Certain Italian soccer players: Controversial cluster
- Lake Mascoma, New Hampshire: Possible cluster linked to cyanobacteria
Mortality and Survival Statistics
Median survival from diagnosis:
- Overall: 20-48 months
- Bulbar onset: 12-26 months
- Limb onset: 24-50 months
Five-year survival rate:
- Overall: 20-30%
- Varies by age, onset type, and care access
Ten-year survival rate:
- Overall: 5-10%
Mortality rate:
- Approximately 1.5-2.5 per 100,000 person-years globally
- Slightly higher in men than women
Demographic Patterns
Age distribution:
- Peak age of onset: 55-75 years
- Juvenile ALS (onset before 25): Approximately 1% of cases
- Late-onset ALS (after 80): Decreasing incidence
Gender ratio:
- Male:Female ratio approximately 1.5:1
- Gender difference decreases with advancing age
- Bulbar-onset more common in women
Ethnic variations:
- Higher rates in Caucasian populations
- Lower rates in Hispanic, African, and Asian populations
- Likely influenced by both genetic and environmental factors
Temporal Trends
Changes over time:
- Apparent increasing incidence in many regions
- May partly reflect improved diagnosis and reporting
- Some evidence for actual increases in certain populations
Age-specific trends:
- Increasing rates in older populations
- Stable rates in younger populations
11. Recent Research & Future Prospects
ALS research is advancing rapidly across multiple fronts:
Recent Research Breakthroughs
Genetic discoveries:
- Identification of new ALS-associated genes (KIF5A, CCNF, TBK1)
- Better understanding of C9orf72 repeat expansion mechanisms
- Development of more comprehensive genetic testing panels
Biomarker development:
- Neurofilament light chain (NfL) in blood and CSF as disease activity marker
- TDP-43 detection methods
- Neuroimaging biomarkers
- Microbiome signatures
Pathophysiology insights:
- RNA metabolism abnormalities
- Nucleocytoplasmic transport defects
- Stress granule dynamics
- Microglial and astrocyte contributions
- Prion-like spread of protein misfolding
Clinical research advances:
- Improved clinical trial design
- Patient stratification approaches
- Remote monitoring technologies
- Updated outcome measures
Current Therapeutic Approaches Under Investigation
Gene-targeted therapies:
- Antisense oligonucleotides for C9orf72, SOD1, and other mutations
- miRNA-based approaches
- Gene replacement strategies
- CRISPR/Cas9 gene editing
Cellular therapies:
- Mesenchymal stem cells
- Neural progenitor cells
- Modified immune cells
- Exosome-based approaches
Small molecule drugs:
- Targeting protein aggregation
- Enhancing autophagy and protein clearance
- Modulating neuroinflammation
- Improving mitochondrial function
Immunotherapies:
- Monoclonal antibodies against misfolded proteins
- Regulatory T-cell approaches
- Modulation of microglial activation
Combination therapies:
- Multi-drug approaches targeting different pathways
- Drug combinations with non-pharmacological interventions
- Sequential therapeutic strategies
Technology and Supportive Care Innovations
Advanced assistive technology:
- Brain-computer interfaces
- Eye-tracking improvements
- Thought-to-speech devices
- Exoskeletons for mobility
- Smart home integration
Respiratory support advances:
- Improved non-invasive ventilation
- Diaphragmatic pacing
- Portable ventilation options
Telemedicine and remote monitoring:
- Home-based clinical trials
- Remote vital capacity monitoring
- Virtual multidisciplinary care
Precision Medicine Approaches
Personalized therapy selection:
- Genetic profile-based treatment
- Biomarker-guided therapy
- Patient-specific iPSC models
Predictive modeling:
- Machine learning for prognosis prediction
- Treatment response modeling
- Computational drug repurposing
Early intervention strategies:
- Presymptomatic therapy for genetic carriers
- Early biomarker-based intervention
- Preventive approaches for high-risk individuals
Clinical Trial Landscape
Novel trial designs:
- Platform trials testing multiple drugs
- Adaptive designs
- Basket trials for genetic subgroups
- Remote decentralized trials
Expanded access programs:
- Increasing availability of investigational treatments
- Right-to-try considerations
- Compassionate use frameworks
Patient-centric research:
- Greater involvement of patients in trial design
- Focus on patient-reported outcomes
- Quality of life as primary endpoint in some trials
Long-Term Outlook
While a complete cure remains elusive, several developments offer hope:
- Multiple therapeutic approaches in late-stage clinical development
- Increasing focus on presymptomatic intervention
- Greater understanding of disease heterogeneity
- Improved supportive care extending survival and quality of life
- Growing research funding and global collaboration
- Potential for significant breakthroughs in gene therapy and cellular approaches
12. Interesting Facts & Lesser-Known Insights
Notable Cases and Unique Presentations
- Stephen Hawking: Lived with ALS for 55 years following diagnosis at age 21, an extraordinarily rare case of longevity with the disease
- Lou Gehrig: Baseball legend whose diagnosis in 1939 brought ALS to public awareness in the United States
- Mao Zedong: Evidence suggests the Chinese leader may have had ALS in his later years
- Jason Becker: Musician who continues to compose music using eye-movement technology despite having ALS for over 30 years
- Steve Gleason: Former NFL player who has become a prominent ALS advocate and technology pioneer
Unusual Disease Characteristics
- Eye movements preserved: Even in advanced stages, most patients retain control of eye muscles
- Sensory functions intact: Touch, pain, temperature sensation, and vision typically remain normal
- Bladder and bowel control: Usually preserved until very late stages
- Regional differences in susceptibility: Different motor neuron pools show varying vulnerability
- Variable progression: Some patients experience rapid decline, while others have long plateaus
- Cognitive spectrum: From no cognitive changes to frontotemporal dementia
Occupational and Environmental Links
- Military service: Veterans have approximately twice the risk of developing ALS
- Professional athletes: Some evidence for higher rates in soccer and American football players
- Blue-collar occupations: Electricians, construction workers, and agricultural workers may have elevated risk
- Cyanobacteria exposure: Potential link to certain algal blooms and ALS clusters
- β-Methylamino-L-alanine (BMAA): Neurotoxin found in certain foods linked to Western Pacific ALS clusters
Research and Funding History
- Ice Bucket Challenge: Viral social media campaign in 2014 raised over $115 million for ALS research
- Lou Gehrig’s speech: His “Luckiest Man” speech at Yankee Stadium in 1939 remains an iconic moment in disease awareness
- MLB connection: Major League Baseball continues annual “Lou Gehrig Day” to raise awareness
- Research funding disparities: Despite similar mortality, ALS receives significantly less research funding than many other diseases
- Patient advocacy impact: ALS patient advocates have successfully influenced FDA policies and drug approvals
Myths and Misconceptions
- “Locked-in” misconception: Many believe ALS patients are completely cognitively intact but trapped in their bodies, yet cognitive changes are relatively common
- Overestimated cognitive preservation: While some patients remain cognitively intact, up to 50% experience some degree of cognitive or behavioral changes
- “No sensory involvement” oversimplification: While primarily a motor disease, subtle sensory abnormalities may occur in some patients
- Treatment misconceptions: Neither riluzole nor edaravone are “cures,” providing modest benefits
- Genetic misunderstanding: Even “sporadic” ALS often has genetic components
Intriguing Scientific Findings
- Split-hand phenomenon: Preferential wasting of specific hand muscles, a distinctive feature of ALS
- Cortical hyperexcitability: Detectable years before symptom onset
- Gut microbiome alterations: Emerging evidence for microbiome differences in ALS patients
- Retinal changes: Subtle changes in the retina detected in some patients, despite no visual symptoms
- TDP-43 pathology: Present in 97% of ALS cases, linking most forms of the disease
Emerging Global Patterns
- Global incidence shifts: Rates potentially changing in developing countries with industrialization
- North-to-south gradient: Higher rates in northern latitudes in both Europe and the United States
- Urban-rural differences: Some evidence for higher rates in rural agricultural regions
- Seasonal patterns: Some studies suggest seasonal variations in ALS onset
- Migration effects: People who migrate adopt the risk of their new country over time
Psychological and Social Aspects
- “ALS personality”: Some research suggests certain personality traits might be more common pre-diagnosis
- Psychological resilience: Many patients show remarkable adaptation and maintained quality of life
- Caregiver impact: Profound effects on family caregivers’ physical and mental health
- Communication evolution: Development of sophisticated methods to maintain connection and expression
- Employment challenges: Working-age patients face complex decisions about disclosure and accommodation
This comprehensive report provides an overview of our current understanding of Amyotrophic Lateral Sclerosis. While scientific understanding continues to evolve rapidly, this report integrates established knowledge with recent discoveries to present a thorough picture of this complex neurodegenerative disease.