⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
Comprehensive Report on Amyotrophic Lateral Sclerosis (ALS)
1. Overview
What is ALS?
Amyotrophic Lateral Sclerosis (ALS), also known as Lou Gehrig’s disease or motor neuron disease (primarily in the UK and Australia), is a progressive neurodegenerative disorder characterized by the selective death of motor neurons in the brain and spinal cord. The term “amyotrophic” refers to the muscle atrophy that results from motor neuron degeneration, while “lateral sclerosis” describes the hardening of the lateral columns of the spinal cord where motor neuron degeneration is prominent.
ALS is a relentlessly progressive condition that leads to paralysis, difficulty speaking, swallowing, and eventually breathing. It is perhaps the most devastating of the neurodegenerative disorders due to its rapid progression while typically preserving cognitive function, meaning patients often remain aware of their physical decline.
Affected Body Parts/Organs
ALS primarily affects the motor neurons, which are nerve cells responsible for voluntary muscle control. Specifically, it impacts:
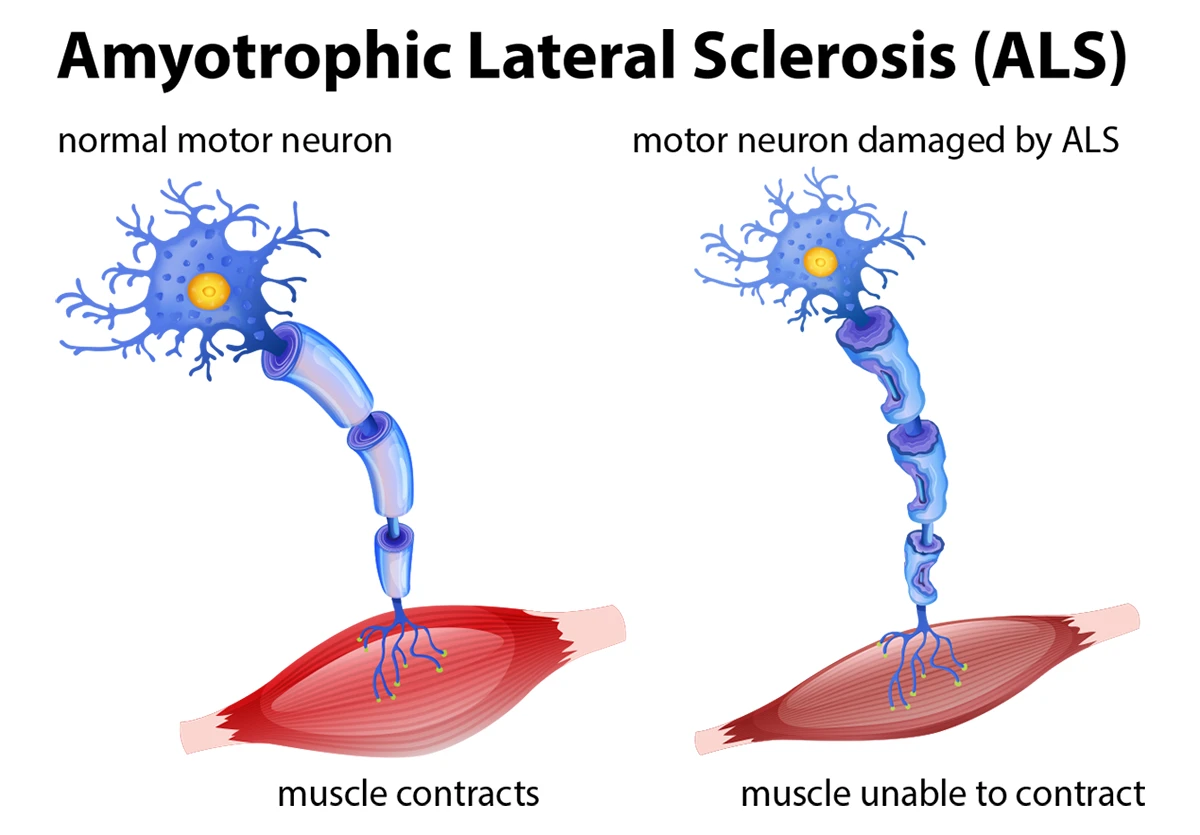
Upper Motor Neurons (UMNs): These originate in the motor cortex of the brain and extend downward to the brainstem and spinal cord. They regulate and fine-tune movement commands.
Lower Motor Neurons (LMNs): These extend from the brainstem and spinal cord to the muscles throughout the body. They directly control muscle contraction.
Skeletal Muscles: As motor neurons die, the muscles they control become weak, begin to atrophy, and develop fasciculations (involuntary twitching).
Respiratory System: Progressive weakness affects the diaphragm and intercostal muscles, leading to respiratory compromise, which is typically the cause of death in ALS.
Bulbar Region: In many cases, ALS affects the bulbar motor neurons that control speaking, swallowing, and breathing, leading to dysarthria (difficulty speaking), dysphagia (difficulty swallowing), and respiratory dysfunction.
While ALS primarily affects the motor system, research has increasingly recognized involvement of non-motor systems in some patients, including mild cognitive changes in approximately 50% of patients and frank frontotemporal dementia in about 15%.
Prevalence and Significance
ALS is relatively rare but represents the most common adult-onset motor neuron disease. Key epidemiological features include:
- Global prevalence: 4-6 cases per 100,000 population
- Incidence: 1-2 new cases per 100,000 individuals annually
- U.S. statistics: Approximately 5,000 new diagnoses each year with about 30,000 Americans living with ALS at any given time
- Lifetime risk: Approximately 1 in 300-400 individuals
- Gender distribution: Slightly more common in men than women (1.5:1 ratio), though this difference narrows with age
- Age of onset: Typically between 40-70 years of age, with a median age of 55
- Juvenile ALS: Rare cases with onset before age 25 (less than 10% of cases)
The significance of ALS extends beyond its prevalence due to several factors:
Profound disability: ALS causes progressive paralysis while cognitive function often remains intact, creating significant physical and psychological challenges.
Economic impact: The estimated annual economic burden in the U.S. alone exceeds $1 billion, with per-patient costs of $250,000-$300,000 annually in advanced stages.
Limited treatment options: Despite decades of research, treatment options remain limited, with only two FDA-approved medications (riluzole and edaravone) showing modest benefits in slowing disease progression.
Rapid progression: The median survival time from symptom onset is 2-4 years, though approximately 10% of patients live 10 years or longer.
Research significance: ALS research has broader implications for understanding neurodegeneration, protein misfolding, and motor system biology.
The devastating nature of ALS, combined with its relatively young age of onset compared to other neurodegenerative diseases, has made it the focus of significant advocacy efforts and research funding initiatives in recent decades.
2. History & Discoveries
First Identification
The history of ALS as a recognized medical condition dates back to the 19th century:
Initial Clinical Descriptions
- 1824: Charles Bell first distinguished between sensory and motor nerves, laying the groundwork for understanding motor neuron diseases.
- 1848: François-Amilcar Aran published descriptions of progressive muscular atrophy, which likely included some cases of what would later be known as ALS.
- 1853: Jean-Martin Charcot’s mentor, Guillaume Duchenne, described a form of progressive muscle weakness that shared features with ALS.
Jean-Martin Charcot’s Discovery
The formal identification of ALS as a distinct disease entity is credited to Jean-Martin Charcot (1825-1893), a renowned French neurologist often called the “father of modern neurology.”
- 1869: Charcot delivered lectures at Salpêtrière Hospital in Paris, where he presented the first comprehensive description of ALS after conducting detailed clinical and pathological studies.
- 1874: Charcot published his definitive paper on “Amyotrophic Lateral Sclerosis,” coining the term and describing the disease’s key clinical and pathological features.
- Charcot’s observations: He astutely connected clinical symptoms with post-mortem findings, noting the characteristic combination of muscle wasting (from lower motor neuron damage) and spasticity (from upper motor neuron damage).
Eponymous Association with Lou Gehrig
ALS gained significant public recognition in the United States when it affected a famous sports figure:
- 1939: Lou Gehrig, the renowned New York Yankees baseball player known as “The Iron Horse,” was diagnosed with ALS.
- July 4, 1939: Gehrig delivered his famous “luckiest man on the face of the earth” farewell speech at Yankee Stadium.
- June 2, 1941: Gehrig died from ALS complications, just two years after diagnosis.
- Legacy: The disease became commonly known as “Lou Gehrig’s disease” in North America, significantly raising public awareness.
Major Discoveries and Breakthroughs
Genetic Discoveries
- 1993: A landmark discovery identified mutations in the SOD1 gene as the first known genetic cause of familial ALS.
- 2006: TDP-43 protein aggregates were identified as a major component of ubiquitinated inclusions in ALS, revolutionizing understanding of the disease pathology.
- 2008-2009: Mutations in TDP-43 (TARDBP) and FUS genes were discovered in familial ALS cases.
- 2011: A hexanucleotide repeat expansion in the C9orf72 gene was identified as the most common genetic cause of both familial ALS and frontotemporal dementia.
- 2014-present: Genome-wide association studies have identified over 30 genes associated with ALS risk, highlighting the genetic heterogeneity of the disease.
Pathophysiological Insights
- 1990s: Excitotoxicity was identified as a potential disease mechanism, leading to the development of riluzole.
- Early 2000s: Protein misfolding and aggregation emerged as a central pathological mechanism, similar to other neurodegenerative diseases.
- 2010s: Studies revealed the importance of RNA processing defects and nucleocytoplasmic transport dysfunction in ALS pathogenesis.
- 2015-present: Recognition of the role of neuroinflammation and glial cells in disease progression, shifting from a neuron-centric to a more holistic view of ALS pathology.
Treatment Milestones
- 1995: Riluzole became the first FDA-approved drug for ALS, showing modest benefits in prolonging survival.
- 2017: Edaravone (Radicava) received FDA approval as the second ALS drug, shown to slow functional decline in a subset of patients.
- 2000s-present: Development of multidisciplinary care approaches, which have demonstrated significant improvements in quality of life and survival.
- 2017-present: Antisense oligonucleotide therapies entered clinical trials for genetic forms of ALS, representing a potential breakthrough in targeted therapy.
Evolution of Medical Understanding
The conceptualization of ALS has undergone significant evolution since Charcot’s initial description:
Changing Disease Definitions
- 1930s-1950s: Recognition of ALS as part of a spectrum of motor neuron diseases, including progressive muscular atrophy and primary lateral sclerosis.
- 1990s: Development of the El Escorial criteria for ALS diagnosis, later revised to the Awaji criteria, standardizing diagnosis for research and clinical practice.
- 2000s: Growing recognition of the overlap between ALS and frontotemporal dementia, leading to the concept of an “ALS-FTD spectrum.”
From Clinical to Molecular Understanding
- 19th century: Purely clinical and pathological descriptions based on post-mortem examinations.
- Mid-20th century: Introduction of electrophysiological techniques (EMG/NCS) allowing for more precise diagnosis.
- Late 20th century: Shift toward understanding cellular pathology, including the role of glutamate excitotoxicity.
- Early 21st century: Focus on molecular and genetic underpinnings, with ALS increasingly viewed as a proteinopathy.
- Present: Integration of multiple levels of understanding, from genetic risk to systems neuroscience, embracing the complexity of ALS as a multifactorial disease.
Changing Treatment Paradigms
- Pre-1990s: Primarily symptomatic management with no disease-modifying options.
- 1990s-2000s: Introduction of riluzole and focus on extending survival.
- 2000s-2010s: Development of multidisciplinary care models, improving quality of life.
- 2010s-present: Growing emphasis on personalized medicine approaches based on genetic and clinical subtypes.
- Current era: Integration of advanced technologies, including brain-computer interfaces, artificial intelligence-guided drug discovery, and genetic therapies.
The historical trajectory of ALS research has accelerated dramatically in recent decades, with more discoveries in the past 30 years than in the previous 150. This progress reflects advances in molecular biology, genetics, and neuroscience, as well as the impact of increased research funding driven by patient advocacy organizations such as the ALS Association and the Ice Bucket Challenge phenomenon of 2014, which raised over $115 million for ALS research.
3. Symptoms
ALS symptoms reflect the progressive degeneration of motor neurons, leading to a characteristic pattern of weakness, atrophy, and functional loss. The symptom presentation and progression can vary significantly between individuals, contributing to diagnostic challenges.
Early Symptoms
The initial manifestations of ALS are often subtle and frequently attributed to more benign conditions, leading to diagnostic delays averaging 9-12 months from symptom onset. Early symptoms typically reflect focal onset in a specific body region:
Limb Onset (70-80% of cases)
- Asymmetric weakness: Usually begins in one limb, most commonly in the hand or foot
- Fine motor difficulties: Trouble with buttons, writing, or picking up small objects
- Foot drop: Difficulty lifting the front part of the foot, causing tripping or a slapping gait
- Muscle cramps: Often occur during physical activity or at rest, particularly at night
- Fasciculations: Visible muscle twitching under the skin, often initially painless
- Exercise intolerance: Premature fatigue with activities that were previously tolerable
- Muscle stiffness: Mild spasticity or perceived stiffness in affected limbs
Bulbar Onset (20-30% of cases)
- Speech changes: Subtle slurring, particularly with certain consonants or when fatigued
- Voice quality changes: Nasality or a “wet” quality to the voice
- Intermittent choking: Especially with liquids or pills
- Excessive saliva: Difficulty managing normal saliva production
- Emotional lability: Inappropriate laughing or crying, disproportionate to emotional state
Less Common Presentations
- Respiratory onset (1-3%): Shortness of breath, especially when lying flat, or morning headaches from nocturnal hypoventilation
- Weight loss: Unexplained modest weight loss may precede other noticeable symptoms
- Cognitive/behavioral changes: Subtle personality changes, apathy, or executive dysfunction in some patients
Advanced-Stage Symptoms
As ALS progresses, symptoms spread from the initial site of onset to involve other body regions, eventually affecting most voluntary muscles:
Motor Symptoms
- Widespread weakness: Progressive weakness in multiple limbs
- Severe atrophy: Visible muscle wasting, including in the tongue and hand intrinsic muscles
- Spasticity: Increased muscle tone and hyperreflexia
- Complete loss of function: Inability to walk, use hands, or perform self-care
- Contractures: Fixed joint deformities from chronic muscle imbalance
- Decubitus ulcers: Pressure sores from immobility
Bulbar Symptoms
- Anarthria: Complete loss of articulate speech
- Severe dysphagia: Inability to swallow safely, requiring feeding tube placement
- Excessive secretions: Difficulty managing saliva and respiratory secretions
- Jaw clonus or jaw fatigue: Difficulty chewing, with involuntary jaw movements in some cases
Respiratory Symptoms
- Dyspnea: Shortness of breath, initially with exertion and later at rest
- Orthopnea: Inability to breathe comfortably when lying flat
- Hypoventilation: Decreased breathing efficiency, leading to carbon dioxide retention
- Weak cough: Inability to clear airway secretions effectively
- Sleep disruption: Sleep-disordered breathing, including sleep apnea
Non-Motor Symptoms
- Cognitive impairment: Executive dysfunction, language deficits, or behavioral changes in 50% of patients
- Frontotemporal dementia: Frank dementia in approximately 15% of cases
- Pain: Secondary pain from immobility, cramps, or spasticity
- Psychological symptoms: Depression, anxiety, or adjustment disorder
- Pseudobulbar affect: Uncontrollable emotional outbursts, often incongruent with true emotions
Common vs. Rare Symptoms
Common Symptoms (>50% of patients)
- Asymmetric limb weakness
- Fasciculations (muscle twitches)
- Hyperreflexia (exaggerated reflexes)
- Dysarthria (slurred speech)
- Dysphagia (swallowing difficulties)
- Muscle atrophy
- Muscle cramps
- Spasticity
- Fatigue
- Weight loss
- Shortness of breath
Uncommon Symptoms (10-50% of patients)
- Cognitive impairment
- Pseudobulbar affect (emotional lability)
- Excessive yawning
- Excessive saliva
- Pain
- Sleep disturbances
- Jaw clonus
- Urinary urgency
- Constipation
Rare Symptoms (<10% of patients)
- Sensory symptoms
- Oculomotor dysfunction (eye movement problems)
- Autonomic dysfunction (blood pressure/heart rate irregularities)
- Parkinsonism features
- Respiratory onset as initial presentation
- Severe dementia
- Sphincter dysfunction
- Pressure sores (in early/mid-stage)
Symptom Progression Over Time
ALS typically follows a pattern of regional spread from the site of onset, though the rate and pattern of progression vary considerably between patients:
Typical Progression Timeline
- Months 0-6: Focal weakness in one region (limb or bulbar), often subtle and asymmetric
- Months 6-12: Spread to adjacent regions, increased disability in the region of onset
- Months 12-24: Involvement of multiple body regions, increasing functional limitations
- Months 24-36: Widespread weakness, typically requiring assistive devices or wheelchairs
- Months 36+: Severe disability, often including loss of speech, swallowing difficulties, and respiratory compromise
Progression Patterns
- Linear progression: Steady decline at a consistent rate (most common)
- Step-wise progression: Periods of stability punctuated by more rapid decline
- Rapid progression: Particularly in bulbar-onset and respiratory-onset cases
- Slow progression: About 10-20% of patients have a more indolent course extending beyond 10 years
Functional Milestones
- Loss of independent ambulation: Median time approximately 12-24 months from diagnosis
- Need for feeding tube: Typically 18-36 months from diagnosis in bulbar-onset cases
- Loss of functional speech: Usually 24-48 months from diagnosis
- Need for ventilatory support: Often 2-4 years from diagnosis, though highly variable
- Death: Median survival 2-4 years from diagnosis, with approximately 10% of patients surviving beyond 10 years
Disease Progression Factors
Several factors influence the rate of symptom progression:
- Age of onset: Younger patients typically progress more slowly
- Site of onset: Bulbar-onset and respiratory-onset cases typically progress more rapidly
- Presence of cognitive impairment: Associated with faster progression
- Genetic factors: Some mutations (e.g., SOD1 A4V) are associated with rapid progression
- Baseline respiratory function: Lower initial vital capacity predicts more rapid progression
- Rate of early progression: The rate of decline in the first 3-6 months often predicts future trajectory
The symptomatic profile of ALS is notable for its remarkable variability between patients while maintaining certain core features that enable diagnosis. This heterogeneity has led to increasing recognition of ALS as a syndrome rather than a single disease entity, with implications for personalized treatment approaches and clinical trial design.
4. Causes
The etiology of ALS is complex and multifactorial, involving a combination of genetic susceptibility, environmental exposures, and cellular mechanisms that contribute to motor neuron degeneration. Despite significant research advances, the precise cause of most ALS cases remains incompletely understood.
Biological Causes
At the cellular and molecular level, several interconnected pathological mechanisms contribute to motor neuron death in ALS:
Protein Misfolding and Aggregation
- TDP-43 pathology: TDP-43 protein mislocalization from the nucleus to cytoplasmic aggregates occurs in approximately 97% of all ALS cases, suggesting a central role in pathogenesis
- SOD1 aggregation: Mutant superoxide dismutase 1 forms toxic protein aggregates in SOD1-mediated familial ALS
- FUS aggregation: Mutations in FUS lead to cytoplasmic mislocalization and aggregation of this RNA-binding protein
- Dipeptide repeat proteins: In C9orf72 mutation carriers, the production of toxic dipeptide repeat proteins contributes to neurodegeneration
- Prion-like spread: Evidence suggests pathological proteins may spread between cells in a prion-like manner, explaining the regional progression of symptoms
RNA Processing Defects
- Splicing abnormalities: Many ALS-associated genes (TDP-43, FUS, hnRNPA1/A2) are involved in RNA splicing, leading to widespread splicing defects
- RNA transport disruption: Impaired RNA trafficking between nucleus and cytoplasm
- Stress granule dynamics: Abnormal stress granule formation and persistence, interfering with normal cellular stress responses
- microRNA dysregulation: Altered microRNA profiles affecting gene expression
Excitotoxicity
- Glutamate dysregulation: Excessive glutamate activity at synapses leads to calcium influx and neuronal damage
- EAAT2 dysfunction: Decreased expression of the astrocytic glutamate transporter EAAT2 (GLT-1) impairs glutamate clearance
- Calcium homeostasis: Disrupted calcium handling in motor neurons increases vulnerability to excitotoxic injury
Mitochondrial Dysfunction
- Energy production deficits: Impaired ATP generation in affected motor neurons
- Calcium buffering failure: Reduced mitochondrial capacity to buffer calcium
- Increased reactive oxygen species: Elevated oxidative stress damages cellular components
- Mitochondrial dynamics: Abnormal mitochondrial transport, fission, and fusion
- Mitochondrial DNA damage: Accumulation of mutations in mitochondrial DNA
Axonal Transport Defects
- Cytoskeletal abnormalities: Disrupted neurofilament organization
- Transport motor dysfunction: Impaired kinesin and dynein function
- Cargo delivery failure: Insufficient transport of mitochondria, vesicles, and other essential materials along the lengthy motor neuron axons
Neuroinflammation
- Microglial activation: Shift from neuroprotective to neurotoxic microglial phenotypes
- Astrogliosis: Reactive astrocytes contributing to disease progression
- Inflammatory cytokines: Elevated levels of pro-inflammatory molecules
- Blood-brain barrier disruption: Allowing peripheral immune cell infiltration
- Complement activation: Inappropriate activation of complement-mediated neuronal damage
Endoplasmic Reticulum (ER) Stress
- Unfolded protein response activation: Chronic ER stress from protein misfolding
- Autophagy dysregulation: Impaired clearance of damaged proteins and organelles
- Vesicular trafficking defects: Disrupted transport between cellular compartments
Genetic and Hereditary Factors
ALS has a significant genetic component, with approximately 5-10% of cases having a clear familial pattern (FALS) and the remainder classified as sporadic (SALS), though genetic factors contribute to both forms:
Major ALS Genes
- C9orf72: Hexanucleotide repeat expansion accounting for approximately 40% of familial ALS and 5-10% of sporadic cases
- SOD1: Over 200 different mutations identified, collectively responsible for 15-20% of familial ALS
- TARDBP (TDP-43): Mutations account for 4-5% of familial ALS
- FUS: Mutations responsible for 4-5% of familial ALS, often associated with juvenile onset
- TBK1: Recently identified gene accounting for 2-3% of familial ALS
- NEK1: Rare variants associated with 3% of both familial and sporadic ALS
- TUBA4A: Cytoskeletal protein gene implicated in approximately 1% of familial ALS
Genetic Risk Factors
- ATXN2: Intermediate-length polyglutamine expansions increase ALS risk
- UNC13A: Single nucleotide polymorphisms associated with survival and disease risk
- VEGF: Specific haplotypes affect vascular endothelial growth factor levels and ALS risk
- CHCHD10: Mutations in this mitochondrial protein gene linked to ALS-FTD spectrum
- Polygenic risk: Combined effect of multiple genetic variants of small individual effect
Heritability Patterns
- Autosomal dominant: Most common inheritance pattern in familial ALS
- Autosomal recessive: Seen in rare juvenile forms
- Incomplete penetrance: Some mutation carriers never develop disease
- Variable expressivity: Same mutation can cause different severity/phenotypes within families
- De novo mutations: New mutations that can cause apparently sporadic cases
- Oligogenic inheritance: Multiple genetic risk factors interacting to cause disease
Environmental Causes and Triggers
Environmental factors likely interact with genetic susceptibility to trigger ALS in many cases:
Established Environmental Associations
- Age: The strongest non-genetic risk factor, with peak onset in the 50s-60s
- Military service: Increased risk, particularly in Gulf War veterans
- Physical activity: Professional athletes, particularly in football and soccer, show increased risk
- Smoking: Consistently associated with 1.5-2x increased risk in multiple studies
Potential Environmental Risk Factors
- Heavy metal exposure: Lead, mercury, selenium, and manganese have been implicated
- Pesticides/herbicides: Agricultural chemicals, particularly organophosphates
- Electric shock: History of significant electrical injury
- Cyanobacteria: Exposure to blue-green algae and their toxin BMAA
- Electromagnetic fields: Controversial association requiring further study
- Air pollution: Emerging evidence for particulate matter exposure
- Head trauma: Traumatic brain injury may increase risk or trigger onset
Triggering Events
Some patients report specific events preceding symptom onset:
- Physical trauma: Injuries sometimes precede symptom recognition
- Intensive exercise: Vigorous unaccustomed exertion reported as a trigger in some cases
- Viral infections: Some cases report onset following significant viral illness
- Surgery: Major operations occasionally precede symptom development
- Psychological stress: Major life stressors sometimes temporally associated with onset
Integration: The Multiple Hit Hypothesis
Current understanding suggests ALS typically requires multiple factors converging to cause motor neuron degeneration:
- Genetic susceptibility: Inherited or spontaneous genetic variants affecting key cellular pathways
- Environmental exposures: Accumulation of toxicants or stressors throughout life
- Age-related vulnerability: Declining repair mechanisms and cellular resilience
- Triggering events: Specific stressors that overwhelm compensatory mechanisms
- Cell-autonomous factors: Intrinsic properties making motor neurons particularly vulnerable
- Non-cell-autonomous factors: Contributions from supporting cells (glia, microglia)
This “multiple hit” model helps explain the variable penetrance of genetic mutations, the late-life onset of most cases, and the phenotypic heterogeneity that characterizes ALS. It also suggests that effective treatment may require addressing multiple pathogenic mechanisms simultaneously.
The complex and multifactorial nature of ALS causation presents both challenges and opportunities for research and treatment development. While a complete understanding of ALS etiology remains elusive, the accelerating pace of discovery provides increasing hope for more effective interventions targeting specific pathogenic mechanisms.
5. Risk Factors
Understanding the risk factors for ALS helps identify individuals who may benefit from closer monitoring, guides research into disease mechanisms, and informs potential preventive strategies. These risk factors span demographic characteristics, genetic predisposition, lifestyle factors, and environmental exposures.
Demographic Factors
Age
- Peak incidence: Between ages 50-75, with median age of onset around 55-65 years
- Age-related risk: Incidence increases with age until approximately age 75, then plateaus or slightly decreases
- Early onset: Approximately 10% of cases begin before age 40, often associated with genetic factors
- Juvenile ALS: Rare cases beginning before age 25, frequently linked to specific genetic mutations (FUS, ALS2)
- Age impact on progression: Older age at onset generally associated with more rapid disease progression
Gender
- Male predominance: Men have approximately 1.5 times higher risk than women
- Gender ratio changes with age: The male-to-female ratio decreases from approximately 2:1 before age 50 to nearly 1:1 after age 70
- Bulbar onset: More common in women, particularly post-menopausal
- Survival difference: Some studies suggest slightly worse prognosis in men
- Hormonal factors: Estrogen may have neuroprotective effects, potentially explaining gender differences
Ethnicity and Geographic Variation
- Global distribution: ALS occurs worldwide but with some geographic variations
- Lower rates: Lower incidence reported in Hispanic, African, and Asian populations compared to Caucasians
- Geographic clusters: Higher than expected prevalence in specific regions:
- Guam and Western Pacific (declining in recent decades)
- Kii Peninsula of Japan
- Western New Guinea
- Parts of Finland
- Continental differences: Higher incidence in North America and Europe compared to Asia and South America
- North-South gradient: Some evidence for decreasing incidence from north to south
Genetic Risk Factors
Family History
- Familial ALS risk: 5-10% of patients report a family history of ALS
- First-degree relative risk: 5-10 times increased risk with affected parent or sibling
- Oligogenic inheritance: Some families show patterns suggesting multiple interacting genetic factors
- Incomplete penetrance: Not all individuals with ALS-causing mutations develop the disease
- Sporadic ALS with genetic component: Up to 10-15% of apparently sporadic cases carry known ALS gene mutations
Specific Genetic Factors
- C9orf72 repeat expansion: Most common genetic cause of ALS, with geographic variability in frequency
- Highest in Finnish and other Northern European populations (21-46% of familial ALS)
- Intermediate in Central/Southern Europe and North America (33-47% of familial ALS)
- Rare in Asian and African populations (<5% of familial ALS)
- SOD1 mutations: Second most common cause, with A4V mutation predominant in North America
- Genetic modifiers: Genes that influence age of onset, disease progression, and survival
- ATXN2 repeat length influences age of onset
- UNC13A variants associated with survival
- APOE ε4 may influence cognitive symptoms
Genetic Risk in Specific Populations
- Ashkenazi Jewish population: Higher rate of specific genetic variants (e.g., SOD1 D90A)
- Finnish population: Elevated C9orf72 mutation frequency
- Sardinian population: Increased incidence potentially linked to genetic founder effects
- Korean/Japanese populations: Higher rates of OPTN mutations
Occupational and Environmental Factors
Occupational Exposures
- Military service: 1.5-2 times increased risk, particularly for Gulf War veterans
- Agriculture: Exposure to pesticides, herbicides, and insecticides
- Heavy metal work: Welding, soldering, and other metal-related occupations
- Electrical work: Possibly related to electric shock exposure
- Professional athletics: Football, soccer players show elevated risk
- NFL players have 4 times higher mortality from ALS
- Italian professional soccer players show 6-fold increased risk
- Physical labor: Some evidence for increased risk with high exertion occupations
Environmental Exposures
- Heavy metals: Lead, mercury, and manganese exposures linked to increased risk
- Pesticides: Organophosphates and other agricultural chemicals
- Solvents: Organic solvents including formaldehyde
- Cyanobacteria: BMAA toxin exposure through water or food sources
- Electromagnetic fields: Controversial association requiring further evidence
- Air pollution: Particulate matter and traffic-related air pollution
- Water quality: β-Methylamino-L-alanine (BMAA) and other waterborne toxins
Lifestyle Factors
Physical Activity
- Intense physical exercise: Possible increased risk with very high levels of exertion
- Professional sports: Particularly contact sports with head trauma
- Mechanism hypotheses:
- Excitotoxicity from repeated high-intensity activity
- Oxidative stress from heightened metabolism
- Subclinical trauma
- Selection bias (genetically predisposed individuals may excel at athletics)
- Controversy: Some studies suggest moderate physical activity may be protective
Smoking
- Consistent association: 1.5-2 times increased risk in current smokers
- Dose-response: Risk correlates with pack-years of smoking
- Persistence: Risk remains elevated for approximately 10 years after quitting
- Possible mechanisms:
- Oxidative stress
- Neurotoxic effects of chemicals in tobacco
- Inflammation
- Impaired vascular function
Body Mass and Metabolic Factors
- Premorbid BMI: Lower premorbid BMI associated with increased risk
- Metabolic rate: Higher physical fitness/metabolic conditioning may correlate with risk
- Lipid profiles: Dyslipidemia may modify disease risk and progression
- Diabetes: Type 1 diabetes potentially associated with increased risk
- Physical structure: Ectomorphic body type (tall, lean) may correlate with increased risk
Pre-existing Medical Conditions
Neurological Conditions
- Frontotemporal dementia: Significant overlap with ALS (15% of ALS patients develop FTD)
- Head trauma: History of traumatic brain injury may increase risk
- Polio: Prior poliomyelitis infection potentially associated with later development of ALS-like syndromes
- Parkinsonism: Occasional co-occurrence, particularly in specific genetic forms
Autoimmune and Inflammatory Conditions
- Other autoimmune diseases: Modest associations with multiple sclerosis, celiac disease, and asthma
- Inflammatory markers: Elevated inflammatory markers may predict risk or progression
- Immune system polymorphisms: Variations in cytokine genes may modify risk
Other Medical Risk Factors
- Viral infections: Some evidence for enterovirus involvement
- Bacterial infections: Emerging research on microbiome alterations
- Metabolic disorders: Potential links to defects in energy metabolism
- Cancer history: Inverse relationship with some cancers (potentially shared protective factors)
Risk Factor Interactions
The etiology of ALS likely involves complex interactions between multiple risk factors:
- Gene-environment interactions: Genetic susceptibility modified by environmental exposures
- Gene-gene interactions: Multiple genetic variants with additive or synergistic effects
- Cumulative exposures: Lifetime accumulation of multiple low-level risk factors
- Critical periods: Developmental windows of heightened vulnerability
- Aging effects: Declining compensatory mechanisms with advancing age
Protective Factors
Some factors appear to be associated with reduced ALS risk:
- Mediterranean diet: Higher intake of antioxidant-rich foods, omega-3 fatty acids
- Vitamin E consumption: Dietary or supplemental intake
- Moderate exercise: Possible protective effect of regular moderate physical activity
- Non-steroidal anti-inflammatory drugs (NSAIDs): Some evidence for protective effect
- Statins: Controversial potential protective effect requiring further study
Understanding ALS risk factors remains an active area of research, with new associations continuing to emerge. The complex interplay between genetic susceptibility and environmental exposures suggests that ALS results from multiple “hits” rather than a single cause, explaining the heterogeneity in presentation, progression, and response to treatments observed among patients.
6. Complications
As ALS progresses, it leads to numerous complications affecting multiple body systems. These complications contribute significantly to morbidity, mortality, and reduced quality of life, requiring proactive management by a multidisciplinary care team.
Respiratory Complications
Respiratory complications are the leading cause of death in ALS, resulting from progressive weakness of the diaphragm and intercostal muscles:
Respiratory Insufficiency
- Hypoventilation: Initially during sleep, progressing to daytime
- Symptoms: Morning headaches, excessive daytime sleepiness, fatigue, poor concentration
- Progression: Gradual decline in forced vital capacity (FVC) and maximum inspiratory pressure (MIP)
- Timing: Typically begins when FVC falls below 50% of predicted
- Monitoring: Regular pulmonary function testing recommended every 3 months
Aspiration Pneumonia
- Mechanism: Dysphagia leading to food or liquid entering the airways
- Incidence: Occurs in 15-25% of patients during the disease course
- Risk factors: Bulbar symptoms, reduced cough strength, immobility
- Consequences: Hospitalization, accelerated decline, significant mortality risk
- Prevention: Feeding tube placement, thickened liquids, postural techniques
Reduced Airway Clearance
- Weak cough: Inability to generate sufficient force to clear secretions
- Measurement: Peak cough flow below 270 L/min indicates high risk
- Consequences: Mucus plugging, atelectasis, secondary infections
- Management: Assisted cough techniques, mechanical insufflation-exsufflation devices
Ventilatory Failure
- End-stage manifestation: Terminal respiratory failure
- Symptoms: Dyspnea, orthopnea, paradoxical breathing, accessory muscle use
- Management options:
- Non-invasive ventilation (NIV)
- Invasive ventilation via tracheostomy
- Palliative approaches to dyspnea management
- Decision points: Advance care planning regarding ventilatory support
Nutritional Complications
Nutritional complications arise from a combination of dysphagia, hypermetabolism, and upper extremity weakness:
Dysphagia
- Prevalence: Eventually affects 80-95% of patients
- Manifestations:
- Difficulty with specific food textures (initially dry or crumbly foods)
- Prolonged meal times
- Frequent choking or coughing while eating
- Wet vocal quality after swallowing
- Consequences: Reduced oral intake, weight loss, social isolation, aspiration
- Assessment: Videofluoroscopic swallow studies, bedside swallow evaluations
Malnutrition
- Weight loss: Occurs in 80% of patients, 5-10% of weight loss in 25-30% of patients at diagnosis
- Mechanisms:
- Reduced intake due to dysphagia
- Hypermetabolism (increased metabolic rate seen in 50-60% of patients)
- Difficulty feeding oneself due to upper extremity weakness
- Consequences:
- Accelerated disease progression
- Reduced respiratory muscle strength
- Immunocompromise
- Pressure ulcer risk
- Fatigue and weakness
- Monitoring: Regular weight checks, body composition analysis
Dehydration
- Causes: Reduced fluid intake due to dysphagia, fear of choking
- Complications: Constipation, thickened secretions, urinary issues
- Management: Alternative hydration strategies, feeding tube
Enteral Nutrition Complications
- Feeding tube placement: Gastrostomy (PEG, RIG, PIG) or nasogastric
- Procedural risks: Higher with declining respiratory function (FVC <50%)
- Post-placement complications:
- Site infections (5-10%)
- Tube displacement or blockage (10-15%)
- Aspiration despite feeding tube (5-10%)
- Gastrointestinal symptoms (bloating, diarrhea)
Communication Complications
Progressive loss of verbal communication ability has profound psychosocial implications:
Dysarthria
- Prevalence: Eventually affects 80-95% of patients
- Progression: From mild slurring to complete anarthria (inability to articulate speech)
- Components:
- Imprecise consonants
- Hypernasality
- Strained-strangled voice quality
- Reduced volume
- Slow rate
- Functional impact: Telephone use difficulties, conversational limitations, professional impacts
Augmentative and Alternative Communication (AAC) Challenges
- Timing issues: Optimal timing for AAC intervention
- Physical limitations: Upper extremity weakness limiting device use
- Eye movement control: Critical for eye-gaze devices, may be affected in advanced stages
- Cognitive issues: May impact ability to learn new systems
- Technology abandonment: Up to 30% of prescribed AAC devices not used long-term
Social Isolation
- Communication barriers: Reduced social participation
- Technology limitations: Difficulty expressing complex thoughts, emotions
- Identity changes: Loss of voice as part of personal identity
- Relationship strain: Changed communication dynamics with family/caregivers
Musculoskeletal Complications
The progressive muscle weakness in ALS leads to several musculoskeletal complications:
Contractures
- Mechanism: Muscle imbalance, reduced movement, and spasticity
- Common locations: Ankles, knees, hips, shoulders, elbows, and hands
- Consequences: Pain, positioning difficulties, hygiene challenges
- Prevention: Regular range of motion exercises, positioning, splinting
Spasticity
- Prevalence: Present in 40-60% of patients, varying in severity
- Impact: Can cause pain, interfere with positioning, and contribute to contractures
- Beneficial aspects: May partially compensate for weakness, enabling standing/transfers
- Management: Physical therapy, medications, focal interventions
Pressure Ulcers
- Risk factors: Immobility, malnutrition, reduced tissue perfusion
- Common locations: Sacrum, heels, greater trochanters, ischial tuberosities
- Prevention: Regular repositioning, pressure-relieving surfaces, skin checks
- Management: Specialized wound care when developed
Falls and Fractures
- Fall risk: Particularly high during transitional mobility phases
- Causes: Weakness, spasticity, equipment misuse
- Consequences: Fractures, head injuries, fear of falling, accelerated decline in mobility
- Prevention: Appropriate assistive devices, environmental modifications, balance training
Psychological and Cognitive Complications
ALS affects mental wellbeing and cognitive function in many patients:
Depression and Anxiety
- Prevalence: Clinical depression in 20-50% of patients
- Anxiety disorders: Panic attacks, generalized anxiety in 30-40%
- Reactive vs. neurologic: Both psychological reactions to diagnosis/progression and direct neurologic components
- Consequences: Reduced quality of life, adherence challenges, increased caregiver burden
- Assessment challenges: Somatic symptoms overlap with ALS symptoms
Cognitive Impairment
- Spectrum:
- Subtle executive dysfunction: 35-40% of patients
- Moderate cognitive impairment: 15-20%
- Frank frontotemporal dementia: 10-15%
- Domains affected:
- Executive function (most common)
- Language
- Social cognition
- Behavior
- Implications:
- Decision-making capacity
- Adherence to recommendations
- Caregiver burden
- Ability to use assistive technology
Pseudobulbar Affect
- Prevalence: 20-50% of patients
- Manifestation: Involuntary laughing or crying incongruent with emotional state
- Impact: Social embarrassment, misinterpretation by others
- Treatment: Specific medications (dextromethorphan/quinidine combination)
Existential Distress
- Common concerns:
- Loss of autonomy
- Becoming a burden
- Fear of suffocation
- Uncertainty about disease trajectory
- Impact: Significant suffering beyond physical symptoms
- Interventions: Psychological support, spiritual care, meaning-centered therapies
Other Systemic Complications
Sleep Disturbances
- Prevalence: 70-80% of patients report poor sleep quality
- Causes:
- Sleep-disordered breathing
- Inability to adjust position
- Cramps and fasciculations
- Depression and anxiety
- Consequences: Fatigue, cognitive impairment, reduced quality of life
Pain
- Prevalence: 50-70% of patients experience significant pain
- Sources:
- Muscle cramps
- Spasticity
- Joint pain from immobility
- Pressure-related discomfort
- Neuropathic elements
- Impact: Reduced quality of life, sleep disturbance, psychological distress
Autonomic Dysfunction
- Manifestations:
- Excessive sweating
- Blood pressure fluctuations
- Temperature dysregulation
- Gastrointestinal motility issues
- Prevalence: Present in 30-50% of patients, often underrecognized
Venous Thromboembolism
- Risk factors: Immobility, dehydration, inflammatory state
- Prevalence: Deep vein thrombosis in 10-15% of patients
- Pulmonary embolism: Significant cause of morbidity and mortality
- Prevention: Controversial due to practical challenges and limited evidence
Long-term Impact and Disability Progression
ALS inevitably leads to progressive disability, but the pattern and timeline vary considerably:
Functional Loss Sequence
The typical sequence of functional loss, though with individual variation:
- Fine motor skills (writing, buttoning)
- Gross motor arm function (lifting, reaching)
- Ambulation (walking, transfers)
- Speech intelligibility
- Swallowing safety
- Head control
- Respiratory sufficiency
Disability Milestones
- Loss of independent ambulation: Median 12-18 months from diagnosis
- Loss of functional arm use: Typically 12-24 months from diagnosis
- Loss of intelligible speech: Variable, but often 18-30 months in bulbar-onset cases
- Need for feeding tube: Often 18-36 months from diagnosis
- Need for ventilatory support: Typically 24-48 months from diagnosis
Mortality
- Median survival: 3-5 years from symptom onset
- Survival variability:
- 20% survive 5+ years
- 10% survive 10+ years
- 5% survive 20+ years
- Common causes of death:
- Respiratory failure (most common)
- Pneumonia
- Cardiovascular events
- Complications of immobility
Factors Influencing Progression
- Phenotypic factors:
- Bulbar onset (faster progression)
- Respiratory onset (fastest progression)
- Pure lower motor neuron (slower progression)
- Age at onset (younger = slower progression)
- Genetic factors:
- SOD1 A4V mutation (rapid progression)
- SOD1 D90A homozygous (slow progression)
- C9orf72 (variable, often with cognitive features)
- Baseline factors:
- Respiratory function at diagnosis
- Nutritional status
- Cognitive status
The numerous complications of ALS underscore the need for comprehensive care delivered by multidisciplinary teams specialized in ALS management. Proactive anticipation and management of these complications can significantly improve quality of life and potentially extend survival, even in the absence of disease-modifying treatments that substantially alter the underlying disease progression.
7. Diagnosis & Testing
Diagnosing ALS presents significant challenges due to its heterogeneous presentation, the absence of a definitive biomarker, and the overlap with other neurological conditions. The diagnostic process typically involves a combination of clinical evaluation, electrodiagnostic studies, laboratory tests, imaging, and sometimes genetic testing.
Diagnostic Process Overview
ALS diagnosis follows a pattern of:
- Recognition of suspicious symptoms
- Exclusion of ALS mimics
- Documentation of upper and lower motor neuron signs
- Observation of progression
- Confirmation through established diagnostic criteria
The process takes substantial time, with an average diagnostic delay of 9-12 months from symptom onset, during which a patient may see 3-5 healthcare providers before receiving the correct diagnosis.
Clinical Evaluation
History Taking
- Initial symptoms: Character, location, and progression pattern
- Timeline: Rate of symptom development and spread
- Functional impact: Activities affected by symptoms
- Associated symptoms: Cognitive changes, sensory symptoms, autonomic features
- Family history: ALS, frontotemporal dementia, or other neurodegenerative diseases
- Occupational history: Exposures to toxins, excessive physical exertion
- Medical history: Pre-existing conditions, medications
Neurological Examination
- Upper motor neuron (UMN) signs:
- Hyperreflexia (exaggerated reflexes)
- Spasticity (increased muscle tone)
- Hoffman’s sign (finger flexor reflex)
- Babinski sign (upgoing plantar response)
- Clonus (rhythmic muscle contractions with stretch)
- Pseudobulbar features (pathological laughing/crying)
- Lower motor neuron (LMN) signs:
- Muscle weakness
- Muscle atrophy
- Fasciculations (visible muscle twitching)
- Hyporeflexia (reduced reflexes in affected segments)
- Flaccidity (reduced muscle tone)
- Distribution assessment:
- Documenting pattern of spreading
- Evidence of contiguous spread
- Bulbar region examination (tongue, facial, pharyngeal muscles)
- Respiratory muscle assessment
Functional Testing
- ALS Functional Rating Scale-Revised (ALSFRS-R):
- 12-item scale measuring functional capacity
- Scores range from 0 (maximum disability) to 48 (normal)
- Decline of approximately 0.9 points/month in typical ALS
- Forced Vital Capacity (FVC):
- Measures respiratory function
- Values <80% suggest respiratory involvement
- Critical decision point at <50% for interventions
- Hand-held dynamometry:
- Quantitative muscle strength testing
- Useful for monitoring progression
- Timed functional tests:
- 9-hole peg test (fine motor)
- Timed up-and-go (mobility)
- Timed speech samples (bulbar function)
Electrodiagnostic Studies
Electrodiagnostic testing is the most important ancillary test in ALS diagnosis:
Electromyography (EMG)
- Purpose: Detect LMN dysfunction and exclude other disorders
- Findings in ALS:
- Fibrillation potentials and positive sharp waves (denervation)
- Fasciculation potentials (spontaneous motor unit discharges)
- Large, long-duration, polyphasic motor unit potentials (reinnervation)
- Reduced recruitment pattern
- Unstable motor units
- Distribution requirement: Evidence of active and chronic denervation in at least three body regions
- Limitations: Normal in very early disease; nonspecific abnormalities in other conditions
Nerve Conduction Studies (NCS)
- Purpose: Rule out peripheral nerve, neuromuscular junction, or muscle disorders
- Expected findings in ALS:
- Normal sensory nerve action potentials
- Normal or mildly reduced compound muscle action potentials
- Normal or mildly reduced conduction velocities
- No conduction blocks (important for excluding multifocal motor neuropathy)
- Importance: Critical for differential diagnosis
Specialized Electrodiagnostic Tests
- Motor unit number estimation (MUNE):
- Quantifies surviving motor units
- Research tool for monitoring progression
- Electrical impedance myography (EIM):
- Measures electrical properties of muscle
- Potential biomarker of disease progression
- Transcranial magnetic stimulation (TMS):
- Assesses upper motor neuron function
- May detect subclinical UMN involvement
Laboratory Testing
While no laboratory test can confirm ALS, several tests help exclude mimics:
Routine Laboratory Tests
- Complete blood count: Rules out anemia, infection
- Comprehensive metabolic panel: Assesses organ function, electrolytes
- Thyroid function tests: Excludes thyroid disorders
- Creatine kinase: Mildly elevated (2-10x normal) in 70-75% of ALS patients
- Erythrocyte sedimentation rate: Rules out inflammatory conditions
- Vitamin B12 and folate levels: Excludes deficiency myelopathy
- Serum protein electrophoresis: Rules out paraproteinemic neuropathies
Specialized Laboratory Tests
- Paraneoplastic antibody panel: When suspicion of paraneoplastic syndrome
- Anti-GM1 ganglioside antibodies: To exclude multifocal motor neuropathy
- Anti-acetylcholine receptor and anti-MuSK antibodies: Rules out myasthenia gravis
- Heavy metal screening: When occupational exposure suspected
- HIV testing: When risk factors present
- Lyme disease serology: In endemic areas with appropriate clinical picture
- Anti-MAG antibodies: For demyelinating neuropathies
- Hexosaminidase A: In patients of Ashkenazi Jewish descent (Tay-Sachs carrier screening)
Cerebrospinal Fluid (CSF) Analysis
- Indications: Atypical presentations, rapid progression, young-onset cases
- Expected findings in ALS:
- Normal cell count and protein
- Mildly elevated protein in 30-40% of cases (usually <100 mg/dL)
- Elevated neurofilament light chain levels (research biomarker)
- Alternative findings: May suggest inflammatory, infectious, or neoplastic conditions
Imaging Studies
Neuroimaging primarily helps exclude ALS mimics rather than confirming the diagnosis:
Magnetic Resonance Imaging (MRI)
- Brain MRI findings:
- Often normal in early ALS
- Hyperintensity of the corticospinal tracts on T2/FLAIR sequences (50-60% of cases)
- Precentral gyrus atrophy in advanced cases
- Frontotemporal atrophy in ALS-FTD spectrum
- Spinal MRI findings:
- Often normal or age-appropriate changes
- Cervical spondylotic myelopathy must be ruled out
- Occasionally shows T2 hyperintensity in the anterior horn region
- Key exclusionary findings:
- Structural lesions (tumors, syrinx)
- Multiple sclerosis-like lesions
- Vascular lesions in relevant areas
- Significant spondylotic changes with cord compression
Advanced Neuroimaging
- Diffusion tensor imaging (DTI):
- Reveals corticospinal tract involvement before conventional MRI
- Research tool, not routinely used clinically
- Functional MRI (fMRI):
- Demonstrates altered motor network activation
- Research applications in understanding compensatory mechanisms
- Magnetic resonance spectroscopy (MRS):
- Shows reduced N-acetylaspartate/creatine ratio in motor cortex
- Potential biomarker for UMN dysfunction
- PET/SPECT imaging:
- Hypometabolism in motor and frontal regions
- Research tool, limited clinical utility
Muscle and Nerve Biopsy
Muscle Biopsy
- Indications: Atypical presentations suggesting myopathy or inflammatory muscle disease
- Findings in ALS:
- Grouped atrophy (denervation pattern)
- Fiber type grouping (reinnervation)
- Angular atrophic fibers
- No inflammatory infiltrates
- Limitations: Invasive, rarely needed for diagnosis
Nerve Biopsy
- Indications: Rarely indicated; considered when sensory symptoms prominent
- Expected findings in ALS: Normal or minimal abnormalities
- Alternative diagnoses: May reveal vasculitis, amyloidosis, or inflammatory neuropathies
Genetic Testing
Indications for Genetic Testing
- Definite family history of ALS: First or second-degree relatives with ALS
- Young-onset cases: Patients under 40 years
- Slow progression patterns: Atypically indolent course
- Geographic/ethnic groups with known mutations: Finnish, Scandinavian (C9orf72)
- Presence of frontotemporal dementia features: Especially with family history of FTD
- Research purposes: With appropriate consent
Common Genetic Tests
- C9orf72 repeat expansion: Most common genetic cause, especially in those with concurrent FTD
- SOD1 sequencing: Second most common genetic cause
- ALS gene panels: Commercially available panels including 30+ ALS-associated genes
- Whole exome/genome sequencing: For research or when high suspicion with negative panels
Genetic Counseling Considerations
- Pre-test counseling: Essential given implications for family members
- Incomplete penetrance: Not all mutation carriers develop disease
- Psychological impact: Testing may cause significant distress
- Reproductive implications: Options for family planning
- Insurance concerns: Potential discrimination issues
Diagnostic Criteria
Several sets of diagnostic criteria have been developed to standardize ALS diagnosis:
El Escorial Criteria (World Federation of Neurology)
Classifies cases based on the certainty of diagnosis:
- Definite ALS: UMN and LMN signs in three regions
- Probable ALS: UMN and LMN signs in two regions with UMN signs rostral to LMN signs
- Probable ALS – Laboratory supported: UMN signs in one or more regions and LMN signs defined by EMG in at least two regions
- Possible ALS: UMN and LMN signs in one region, or UMN signs in at least two regions, or UMN and LMN signs in two regions with no UMN signs rostral to LMN signs
- Suspected ALS: Pure LMN syndrome
Awaji Criteria
Modified El Escorial criteria giving equal weight to clinical and electrophysiological evidence of LMN dysfunction, increasing diagnostic sensitivity without reducing specificity.
Gold Coast Criteria (2020)
Newest criteria simplifying diagnosis to:
- Clinically definite ALS: Progressive UMN and LMN dysfunction in at least one limb or region
- Clinically probable ALS: Progressive UMN and LMN dysfunction in any region, and the diagnosis is supported by neurophysiologically definite changes in one or more regions
Diagnostic Challenges and Pitfalls
ALS Mimics
Conditions frequently mistaken for ALS include:
- Cervical spondylotic myelopathy: Compressive cervical spine disease
- Multifocal motor neuropathy with conduction block: Treatable immune-mediated condition
- Inclusion body myositis: Progressive muscle disorder with selective pattern
- Kennedy’s disease (SBMA): X-linked trinucleotide repeat disorder
- Post-polio syndrome: New weakness decades after polio infection
- Myasthenia gravis: Fluctuating weakness with fatigue
- Peripheral neuropathies: Various types affecting motor nerves
- Primary lateral sclerosis: Pure UMN disorder with slower progression
- Progressive muscular atrophy: Pure LMN variant with better prognosis
- Adult polyglucosan body disease: Rare glycogen storage disorder
Misdiagnosis Statistics
- Initial misdiagnosis rate: 25-40% of ALS patients
- Common initial diagnoses: Cervical radiculopathy, peripheral neuropathy, multiple sclerosis
- Mean time to correct diagnosis: 10-16 months from symptom onset
- Consequences: Delayed appropriate care, unnecessary procedures, psychological impact
Early Detection Methods and Efficacy
Current approaches to improve early diagnosis include:
- Screening tools for primary care: Questionnaires to identify suspicious symptoms
- EMG protocols: Standardized comprehensive protocols for suspected cases
- Biomarker development:
- Neurofilament light chain (NfL) in blood/CSF: 85-90% sensitivity
- MicroRNA panels: Emerging potential
- Proteomics approaches: In development
- Genetic risk profiling: Polygenic risk scores in research setting
- Specialist ALS clinics: Improved diagnostic accuracy and reduced delays
Despite these approaches, early diagnosis remains challenging, with limited progress in reducing diagnostic delay over the past two decades. The most effective current approach combines careful clinical assessment by experienced neurologists with appropriate electrodiagnostic testing and targeted investigation to exclude mimics.
The diagnostic process for ALS requires a systematic approach, patience, and experience. Given the devastating nature of the diagnosis and lack of curative treatments, physicians must balance the need for thorough investigation against the anxiety of prolonged diagnostic uncertainty. Ongoing research into biomarkers and improved diagnostic criteria aims to facilitate earlier, more accurate diagnosis, potentially allowing earlier intervention with disease-modifying therapies as they become available.
8. Treatment Options
Despite significant research efforts, treatment options for ALS remain limited. Current approaches focus on slowing disease progression, managing symptoms, and optimizing quality of life through a multidisciplinary approach.
Disease-Modifying Therapies
Only a few medications have demonstrated modest benefits in slowing ALS progression:
Riluzole
- Mechanism: Glutamate antagonist, reduces excitotoxicity
- Efficacy: Extends survival by approximately 2-3 months on average
- Dosage: 50 mg twice daily
- Administration: Oral tablet; oral suspension (Tiglutik) and oral film (Exservan) formulations available for patients with dysphagia
- Side effects:
- Elevated liver enzymes (monitor LFTs)
- Fatigue
- Nausea
- Dizziness
- Limitations: Modest effect size, cost (though generic versions available)
- Guidelines: Standard of care; recommended for all patients without contraindications
Edaravone (Radicava)
- Mechanism: Free radical scavenger, reduces oxidative stress
- Efficacy: Slows functional decline by approximately 33% in a subset of patients with early-stage, rapidly progressive disease
- Dosage: 60 mg daily for 14 days, followed by 14-day drug-free period
- Administration:
- Originally IV infusion only
- Oral formulation (Radicava ORS) approved in 2022
- Side effects:
- Bruising
- Gait disturbances
- Headache
- Infusion site reactions (IV formulation)
- Limitations:
- Complex administration schedule
- Restrictive initial approval criteria
- High cost
- Limited evidence for broader ALS population
- Guidelines: Generally recommended, though patient selection criteria vary between countries
Sodium Phenylbutyrate-Taurursodiol (Relyvrio/AMX0035)
- Mechanism: Combination therapy targeting endoplasmic reticulum stress and mitochondrial dysfunction
- Efficacy:
- Slows functional decline
- Potential survival benefit of approximately 6-10 months based on extension study data
- Approval status: FDA-approved in 2022
- Administration: Oral suspension, twice daily
- Side effects:
- Gastrointestinal symptoms (diarrhea, abdominal pain)
- Bitter taste
- Respiratory symptoms
- Limitations:
- Relatively new, longer-term data still emerging
- High cost
- Mixed interpretations of efficacy data
- Guidelines: Recently incorporated into treatment algorithms
Masitinib
- Mechanism: Tyrosine kinase inhibitor with anti-inflammatory effects
- Status: Under regulatory review, positive Phase 3 data
- Target population: Patients with moderate disease progression
- Administration: Oral tablets
- Potential side effects: Edema, rash, gastrointestinal symptoms
- Evidence quality: Controversy regarding strength of clinical data
Symptomatic Treatments
Managing symptoms is a crucial aspect of ALS care, significantly impacting quality of life:
Spasticity Management
- Pharmacological approaches:
- Baclofen: Most commonly used, 10-25 mg 3-4 times daily
- Tizanidine: Alternative option, 2-8 mg 3 times daily
- Dantrolene: Less commonly used due to hepatotoxicity risk
- Benzodiazepines: Effective but can cause sedation and respiratory depression
- Interventional approaches:
- Intrathecal baclofen pump: For severe, intractable spasticity
- Botulinum toxin injections: For focal spasticity
- Physical approaches:
- Stretching programs
- Positioning
- Splinting
Secretion Management
- Thick secretions:
- Mechanical insufflation-exsufflation (cough assist) devices
- Mucolytics (guaifenesin, N-acetylcysteine)
- Adequate hydration
- Nebulized saline
- Excess saliva (sialorrhea):
- Anticholinergics (glycopyrrolate, amitriptyline, atropine drops)
- Botulinum toxin injections to salivary glands
- Radiation therapy to salivary glands (in severe cases)
- Suction devices
Pseudobulbar Affect
- Pharmacological:
- Dextromethorphan/quinidine (Nuedexta): FDA-approved, highly effective
- Antidepressants (SSRIs, TCAs): Alternative options
- Non-pharmacological:
- Patient and caregiver education
- Behavioral techniques
Pain Management
- Medication approaches:
- NSAIDs: For musculoskeletal pain
- Gabapentin/pregabalin: For neuropathic elements
- Tricyclic antidepressants: For mixed pain syndromes
- Opioids: For severe, refractory pain
- Non-pharmacological approaches:
- Physical therapy
- Massage
- Heat/cold therapy
- Proper positioning
- Assistive devices
Fatigue Management
- Pharmacological:
- Modafinil: 100-200 mg daily
- Amantadine: 100 mg twice daily
- Non-pharmacological:
- Energy conservation techniques
- Activity pacing
- Assistive devices
- Treatment of contributory factors (depression, sleep disorders)
Sleep Disorders
- Interventions:
- Treatment of underlying causes (respiratory insufficiency, pain)
- Sleep hygiene education
- Positional devices
- Medications (zolpidem, temazepam) with caution regarding respiratory function
Cognitive and Behavioral Symptoms
- Pharmacological approaches:
- SSRIs for depression and anxiety
- Atypical antipsychotics for behavioral symptoms in ALS-FTD (with caution)
- Non-pharmacological:
- Cognitive-behavioral therapy
- Caregiver education and support
- Environmental modifications
- Communication strategies
Nutritional Management
Nutritional support is critical as dysphagia develops and metabolic demands increase:
Dietary Modifications
- Texture modifications:
- Progressive adaptation (soft, minced, pureed)
- Thickened liquids
- Caloric requirements:
- Generally increased (125-150% of predicted)
- Account for hypermetabolism present in many patients
- Protein requirements:
- Typically increased (1.2-1.5 g/kg/day)
- Consideration of renal function
- Supplementation:
- High-calorie oral supplements
- Vitamins D and E (some evidence for benefit)
- Creatine monohydrate (controversial)
Enteral Nutrition
- Timing considerations:
- Weight loss >10% of body weight
- Significant dysphagia with risk of aspiration
- Meal times exceeding 45 minutes
- FVC >50% preferred for procedure safety
- Feeding tube options:
- Percutaneous endoscopic gastrostomy (PEG)
- Radiologically inserted gastrostomy (RIG)
- Per-oral image-guided gastrostomy (PIG)
- Nasogastric tube (temporary solution)
- Management:
- Formula selection based on metabolic needs
- Feeding schedules (bolus vs. continuous)
- Complication prevention and management
Respiratory Support
Respiratory management is crucial as respiratory muscle weakness progresses:
Non-invasive Ventilation (NIV)
- Indications:
- Symptoms of respiratory insufficiency
- FVC <50% predicted
- Maximum inspiratory pressure <60 cm H₂O
- Abnormal nocturnal oximetry
- Elevated pCO₂ >45 mmHg
- Benefits:
- Extends survival by 7-12 months on average
- Improves quality of life and sleep
- Reduces hospitalizations
- Types:
- Bi-level positive airway pressure (BiPAP) – most common
- Volume-assured pressure support
- Mouthpiece ventilation (daytime option)
- Implementation challenges:
- Adaptation period
- Mask interface selection and fitting
- Cognitive impairment affecting compliance
- Bulbar symptoms causing air leakage
Invasive Ventilation
- Considerations:
- Long-term survival possible but with complete dependence
- High financial and caregiver burden
- Advance care planning essential before crisis
- Tracheostomy ventilation:
- 24-hour care requirements
- Communication challenges (may require eye-tracking systems)
- Long-term institutional care often necessary
- International variations:
- Higher rates in Japan (approximately 30%)
- Lower rates in Europe and North America (1-5%)
Secretion Management
- Assisted cough techniques:
- Manual assisted cough
- Mechanical insufflation-exsufflation (MIE)
- Threshold values: peak cough flow <270 L/min
- Airway suctioning:
- Training for caregivers
- Portable suction devices
Multidisciplinary Care
The comprehensive care team approach has been shown to extend survival and improve quality of life:
Multidisciplinary ALS Clinic Components
- Core team members:
- Neurologist (preferably with ALS expertise)
- Nurse coordinator
- Physical therapist
- Occupational therapist
- Speech-language pathologist
- Respiratory therapist
- Nutritionist/dietitian
- Social worker
- Extended team:
- Pulmonologist
- Gastroenterologist
- Palliative care specialist
- Psychologist
- Genetic counselor
- Assistive technology specialist
- Orthotist
Care Coordination
- Clinic model:
- Regularly scheduled visits (typically every 3 months)
- Coordinated same-day evaluations
- Interdisciplinary communication
- Proactive problem-solving
- Community support:
- Home health services
- Home adaptations
- Durable medical equipment provision
- Caregiver training
Benefits of Specialized ALS Care
- Outcomes:
- 7-12 month survival advantage with multidisciplinary care
- Fewer hospitalizations
- Improved quality of life measures
- Better symptom management
- Increased use of adaptive equipment
- Improved end-of-life care
Rehabilitation Approaches
Rehabilitation focuses on maintaining function, preventing complications, and adapting to progressive weakness:
Physical Therapy
- Exercise recommendations:
- Moderate intensity, avoid exhaustion
- Typically resistance exercise at 40-60% maximum
- Combined with adequate rest periods
- Emphasis on fall prevention
- Interventions:
- Range of motion exercises
- Assistive device prescription and training
- Transfer training
- Gait training
- Fall prevention strategies
Occupational Therapy
- Focus areas:
- Activities of daily living adaptation
- Upper extremity orthoses
- Home modification recommendations
- Energy conservation techniques
- Environmental control systems
- Adaptive equipment:
- Built-up utensils
- Button hooks
- Toilet and bathroom adaptations
- Mobile arm supports
Speech-Language Pathology
- Communication interventions:
- Augmentative and alternative communication (AAC) systems
- Voice banking before speech loss
- Speech-generating devices
- Eye-tracking technology for advanced disease
- Training in use of communication devices
- Swallowing interventions:
- Swallowing strategies
- Texture modifications
- Positioning techniques
- Swallowing exercises when appropriate
Psychosocial Support
Psychological and social support is essential for both patients and caregivers:
Psychological Interventions
- Individual counseling:
- Adjustment to diagnosis
- Coping strategies
- End-of-life concerns
- Group support:
- Peer-led support groups
- Online communities
- Shared experience benefits
- Family support:
- Caregiver stress management
- Relationship changes
- Family counseling
Social Services
- Financial planning:
- Disability benefits
- Insurance navigation
- Financial assistance programs
- Legal planning:
- Advance directives
- Power of attorney
- Will preparation
- Community resources:
- ALS Association and other organizations
- Equipment loan programs
- Respite care options
Palliative Care
Palliative care focuses on quality of life and symptom management throughout the disease course:
Early Integration
- Benefits:
- Improved symptom control
- Better advance care planning
- Reduced emergency hospitalizations
- Support for psychological adaptation
- Timing:
- Ideally from diagnosis onwards
- Not limited to end-of-life care
End-of-Life Care
- Hospice services:
- Specialized care team
- Home-based or inpatient options
- Family support
- Bereavement services
- Symptom management:
- Dyspnea management (opioids, anxiolytics)
- Terminal secretion management
- Anxiety and agitation control
- Comfort-focused care
Emerging and Experimental Treatments
Numerous experimental approaches are under investigation:
Gene Therapy
- Antisense oligonucleotides (ASOs):
- Tofersen for SOD1 ALS (FDA-approved under accelerated approval in 2023)
- C9orf72-targeting ASOs in trials
- ATXN2-targeting ASOs to modify TDP-43 toxicity
- Gene replacement:
- AAV-delivered SOD1 silencing
- AAV-mediated gene replacement for genetic forms
Stem Cell Approaches
- Neural stem cell transplantation:
- Multiple clinical trials completed with mixed results
- Potential mechanism: trophic support rather than replacement
- Mesenchymal stem cells:
- Immunomodulatory effects
- Delivery methods: intrathecal, intravenous, intramuscular
- Several trials ongoing
Immunomodulatory Therapies
- NP001 (sodium chlorite):
- Targets neuroinflammation
- Mixed results in clinical trials
- Tocilizumab:
- IL-6 receptor antagonist
- Targets neuroinflammation
- Fingolimod:
- Sphingosine-1-phosphate receptor modulator
- Immune cell trafficking inhibition
Neuroprotective Approaches
- Ibudilast (MN-166):
- Phosphodiesterase inhibitor with anti-inflammatory effects
- Phase 2/3 trials ongoing
- Tauroursodeoxycholic acid (TUDCA):
- Bile acid with anti-apoptotic effects
- Phase 3 trials in progress
- Metformin:
- Multiple mechanisms including AMPK activation
- Repurposed drug in clinical trials
Protein Clearance Enhancement
- Arimoclomol:
- Heat shock protein co-inducer
- Enhances protein folding and clearance
- Phase 3 trial completed with negative results
- Copper-ATSM:
- Copper delivery compound
- Affects SOD1 processing
- Early-phase trials ongoing
Clinical Trial Innovations
- Platform trials:
- Multiple treatment arms against shared placebo
- Adaptive design with interim analyses
- Example: HEALEY ALS Platform Trial
- Remote assessment:
- Decentralized trials with reduced participant burden
- Digital biomarkers for more frequent assessment
- Expanded access to research participation
The treatment landscape for ALS continues to evolve, with increasing emphasis on personalized approaches based on genetic and phenotypic characteristics. While current FDA-approved therapies offer modest benefits, the robust pipeline of investigational agents provides hope for more effective disease-modifying treatments in the future. Meanwhile, multidisciplinary symptomatic management remains the cornerstone of care, significantly impacting quality of life and potentially extending survival.
9. Prevention & Precautionary Measures
Unlike some neurodegenerative diseases where clear preventive strategies exist, ALS prevention remains challenging due to its complex and multifactorial etiology. Current approaches focus primarily on risk reduction, early intervention, and management of modifiable risk factors.
Primary Prevention Possibilities
Primary prevention aims to prevent disease onset entirely. For ALS, evidence-based strategies are limited, but several approaches show potential:
Lifestyle Factors
Smoking cessation:
- Strongest modifiable risk factor (1.5-2 times increased risk)
- Public health campaigns targeting smoking reduction
- Comprehensive tobacco control policies
- Individual counseling and pharmacotherapy for cessation
Physical activity moderation:
- Avoiding excessive high-intensity exercise, particularly in susceptible individuals
- Balanced approach to physical fitness
- Potential risk reduction through moderate regular activity
- Sports safety measures, particularly for contact sports with head injury risk
Dietary considerations:
- Mediterranean diet pattern associated with possible risk reduction
- Adequate vitamin E intake (15 mg/day) from food sources
- Omega-3 fatty acid consumption
- Potential benefits of antioxidant-rich foods
- Maintaining healthy weight (avoiding both obesity and low BMI)
Environmental Exposure Reduction
Occupational safety measures:
- Proper protective equipment for those working with heavy metals
- Reducing pesticide exposure in agricultural settings
- Workplace safety standards for toxin exposure
- Regular monitoring of high-risk occupations
Lead and heavy metal exposure reduction:
- Public water supply monitoring and filtration
- Lead abatement in older buildings
- Consumer product regulations
- Biological monitoring for those at risk
Electromagnetic field exposure:
- While evidence is inconclusive, prudent avoidance of high-level exposures
- Occupational safety standards for electrical workers
- Research into potential mechanisms and thresholds
Military Service Considerations
Deployment health monitoring:
- Screening for early neurological symptoms in veterans
- Research into specific exposures during deployment
- Registry programs for tracking long-term health outcomes
Traumatic brain injury prevention:
- Improved helmet design and use
- Blast exposure mitigation strategies
- Enhanced protocols for TBI assessment and management
Secondary Prevention Approaches
Secondary prevention focuses on early detection and intervention in high-risk individuals:
Genetic Counseling and Testing
Family-focused approaches:
- Identification of at-risk family members
- Genetic counseling regarding inheritance patterns
- Discussion of reproductive options
- Psychological support for living with genetic risk
Genetic testing considerations:
- Presymptomatic testing offered with appropriate counseling
- Testing protocols similar to those used for Huntington’s disease
- Comprehensive pre- and post-test counseling
- Privacy and insurance discrimination protections
Monitoring High-Risk Individuals
Clinical surveillance:
- Regular neurological examinations for at-risk individuals
- Baseline and periodic electrodiagnostic studies in some cases
- Monitoring for subtle early signs (fasciculations, mild weakness)
Biomarker development:
- Blood neurofilament light chain (NfL) monitoring
- Specialized neuroimaging in research settings
- Emerging fluid biomarkers (microRNAs, metabolomics)
Pre-symptomatic Intervention
Research initiatives:
- Pre-symptomatic treatment trials in genetic mutation carriers
- Examples include PREVENT-ALS for SOD1 carriers
- Similar approach to Alzheimer’s prevention trials
Monitoring for triggering factors:
- Avoiding extreme physical or environmental stressors
- Managing inflammatory conditions
- Optimizing general health parameters
Tertiary Prevention
Tertiary prevention aims to reduce the impact of established disease:
Early Diagnosis and Treatment
Reducing diagnostic delay:
- Education of primary care and general neurologists about early signs
- Streamlined referral pathways to specialized centers
- Implementation of screening tools in at-risk populations
Early intervention:
- Prompt initiation of approved therapies
- Early multidisciplinary care involvement
- Proactive symptom management
Disease Course Modification
Comorbidity management:
- Optimal control of diabetes, hypertension, and other conditions
- Minimizing polypharmacy
- Prevention of secondary complications
Nutrition optimization:
- Early nutritional support before significant weight loss
- Proactive monitoring of body composition
- Appropriate caloric and protein intake
Respiratory optimization:
- Early pulmonary function monitoring
- Timely initiation of non-invasive ventilation
- Respiratory muscle training in early stages
Precautionary Measures for Specific Populations
For Individuals with Family History
Healthy lifestyle practices:
- Balanced diet rich in antioxidants
- Regular moderate exercise
- Smoking avoidance
- Stress management techniques
Environmental considerations:
- Occupation selection awareness
- Minimizing toxin exposure
- Consideration of geographic factors in living choices
Participation in research:
- Longitudinal family studies
- Biomarker development research
- Genetic modifier identification
For High-Risk Occupations
Military personnel:
- Post-deployment health assessments
- Long-term veteran health monitoring
- Education about early neurological symptoms
Athletes in contact sports:
- Head injury prevention and management
- Consideration of retirement timing in professional athletes
- Monitoring for early neurological symptoms
Agricultural workers:
- Pesticide safety measures
- Proper use of protective equipment
- Regular health screenings
Industrial workers:
- Heavy metal exposure precautions
- Workplace safety standards adherence
- Air quality monitoring
Preventive Clinical Trials and Research
Current Prevention-Focused Trials
Pre-symptomatic intervention trials:
- Tofersen in pre-symptomatic SOD1 mutation carriers
- Antisense oligonucleotides for C9orf72 carriers
- Various approaches in familial ALS
Risk reduction studies:
- Investigation of vitamin E supplementation
- Evaluation of anti-inflammatory agents
- Studies of metabolic modulators
Epidemiological Research
Population-based registries:
- National ALS registries (e.g., National ALS Registry in the U.S.)
- Global collaborative databases
- Identification of geographical and temporal trends
Risk factor identification:
- Case-control studies of environmental exposures
- Prospective cohort studies in high-risk populations
- Twins studies exploring gene-environment interactions
Preventive Biomarker Development
Imaging biomarkers:
- Advanced MRI techniques detecting pre-symptomatic changes
- PET imaging of neuroinflammation
- Functional connectivity assessment
Fluid biomarkers:
- Blood and CSF neurofilament levels
- TDP-43 detection methods
- Inflammatory marker profiles
- MicroRNA signatures
Public Health Approaches
Awareness and Education
Professional education:
- Training for primary care physicians on early recognition
- Continuing education for neurologists on advances
- Allied health professional awareness of multidisciplinary approaches
Public awareness campaigns:
- Focusing on early symptom recognition
- Highlighting the importance of specialized care
- Addressing misconceptions about the disease
Environmental Health Policies
Regulatory approaches:
- Pesticide and herbicide use regulations
- Heavy metal exposure standards
- Occupational safety guidelines
Research funding:
- Support for environmental factor investigations
- Development of prevention strategies
- Translational research on risk reduction
Future Directions in Prevention
Precision Prevention Approaches
Polygenic risk scores:
- Identification of individuals with heightened genetic susceptibility
- Customized preventive strategies based on genetic profile
- Integration with environmental risk assessment
Personalized risk reduction:
- Tailored interventions based on individual risk factor profile
- Targeted monitoring of highest-risk individuals
- Stratified approach to preventive clinical trials
Novel Preventive Targets
Microbiome modulation:
- Emerging evidence for gut-brain axis relevance
- Potential for probiotic or prebiotic interventions
- Manipulation of metabolites affecting neuroinflammation
Cellular resilience enhancement:
- Approaches targeting protein homeostasis
- Methods to enhance mitochondrial function
- Interventions supporting DNA repair mechanisms
Preventive immunomodulation:
- Targeting neuroinflammatory processes before symptom onset
- Vaccines against protein aggregates (experimental)
- Early intervention in inflammatory cascades
Technological Innovations
Wearable monitoring:
- Detection of subtle motor changes before clinical symptoms
- Continuous physiological monitoring
- Integration with healthcare systems for early alerts
Remote assessment platforms:
- Enabling broader monitoring of at-risk populations
- Frequent, low-burden evaluations
- Digital biomarkers of preclinical changes
While complete prevention of ALS remains challenging given our current understanding of its causes, significant opportunities exist for risk reduction and early intervention. The future of ALS prevention will likely involve a combination of targeted approaches for high-risk individuals and broader public health strategies addressing modifiable risk factors. As our understanding of ALS pathogenesis improves, particularly regarding genetic and environmental interactions, more effective preventive strategies may emerge, potentially including personalized approaches based on individual risk profiles.
10. Global & Regional Statistics
Amyotrophic Lateral Sclerosis demonstrates notable epidemiological patterns and variations across different regions of the world. These patterns provide valuable insights into disease etiology, risk factors, and healthcare disparities.
Global Incidence and Prevalence
Overall Incidence
- Global average incidence: 1.68-2.08 per 100,000 person-years
- Range of reported incidence: 0.3-3.6 per 100,000 person-years, depending on region and methodology
- Annual new cases worldwide: Approximately 100,000-120,000
- Temporal trends: Generally stable or slightly increasing over recent decades in most regions
Overall Prevalence
- Global average prevalence: 4.1-8.4 per 100,000 population
- Range of reported prevalence: 1.0-10.3 per 100,000 population
- Estimated cases worldwide: 350,000-450,000 people living with ALS
- Prevalence-to-incidence ratio: Approximately 3:1, reflecting the relatively short survival duration
Age-Related Patterns
- Age-specific incidence:
- Rare before age 40 (<5% of cases)
- Increases dramatically after age 40
- Peaks between ages 60-75
- Slight decline after age 80, possibly due to competing mortality causes
- Median age at diagnosis:
- Global median: 65 years
- Range: 58-68 years across different regions
- Earlier onset in some genetic forms and specific populations
Gender Distribution
- Male-to-female ratio:
- Overall: 1.2-1.5:1
- Before age 50: Approximately 2:1
- After age 70: Nearly 1:1
- Regional variations:
- Higher male predominance in some Asian populations (up to 3:1)
- More equal gender distribution in Scandinavian countries
- Phenotypic differences:
- Bulbar-onset more common in women
- Earlier age of onset typically observed in men
Regional Differences in Incidence and Prevalence
North America
- United States:
- Incidence: 1.7-2.3 per 100,000 person-years
- Prevalence: 5.0-6.4 per 100,000 population
- Approximately 5,000-6,000 new diagnoses annually
- Estimated 20,000-30,000 Americans living with ALS
- Registry data suggests possible geographic clusters in the Midwest and Northeast
- Canada:
- Incidence: 2.0-2.4 per 100,000 person-years
- Prevalence: 6.7-8.4 per 100,000 population
- Higher rates in eastern provinces compared to western
Europe
- Western Europe:
- Incidence: 2.1-3.6 per 100,000 person-years
- Prevalence: 5.4-10.3 per 100,000 population
- Highest reported incidence globally, particularly in Scandinavia and Ireland
- Eastern Europe:
- Incidence: 1.0-1.8 per 100,000 person-years
- Prevalence: 3.5-5.6 per 100,000 population
- Lower rates compared to Western Europe, though methodology differences exist
- Notable national variations:
- Finland: Particularly high rates (3.6 per 100,000 person-years)
- Ireland: High incidence (2.8 per 100,000 person-years)
- Italy: North-south gradient with higher rates in northern regions
- UK: Regional variations with higher rates in Scotland
Asia
- East Asia:
- Japan: Incidence 1.2-2.3 per 100,000 person-years; prevalence 3.9-6.7 per 100,000
- China: Incidence 0.3-0.8 per 100,000 person-years; prevalence 1.2-3.8 per 100,000
- South Korea: Incidence 1.0-1.5 per 100,000 person-years
- South Asia:
- India: Limited data, estimated incidence 0.2-0.6 per 100,000 person-years
- Notable regional clusters reported in specific regions (e.g., Kii Peninsula in Japan)
- General pattern: Lower rates compared to European populations, though increasing with improved detection
Australia and New Zealand
- Australia:
- Incidence: 1.5-2.3 per 100,000 person-years
- Prevalence: 6.0-8.5 per 100,000 population
- Rates comparable to North American and European populations
- New Zealand:
- Incidence: 1.7-2.2 per 100,000 person-years
- Higher rates in populations of European descent compared to Māori and Pacific Islander populations
Africa
- Limited data available:
- Few population-based studies
- Estimated incidence 0.3-0.8 per 100,000 person-years based on available data
- Likely underdiagnosis and underreporting
- South Africa:
- More data available than other African regions
- Lower reported rates compared to European populations
- Racial differences noted with higher rates in white South Africans
Latin America
- Overall statistics:
- Incidence: 0.7-1.5 per 100,000 person-years
- Prevalence: 2.0-4.5 per 100,000 population
- Regional examples:
- Uruguay: Higher rates, comparable to Southern European populations
- Brazil: Variable rates with higher incidence in southern regions
- Mexico: Lower reported rates, possibly reflecting healthcare access issues
- Admixed populations: Some evidence for lower rates in populations with higher Indigenous American ancestry
Geographic Clusters and Special Populations
Western Pacific Foci
- Guam:
- Historically very high incidence (50-100 times global average) in the Chamorro population during 1950s-1960s
- Dramatic decline since then, now approaching global averages
- Theories include environmental toxins (cycad consumption, BMAA)
- Kii Peninsula (Japan):
- Elevated ALS incidence (2-3 times Japanese average)
- Often accompanied by parkinsonism and dementia
- Ongoing research into environmental and genetic factors
Sardinia, Italy
- Elevated incidence: 2.5-3.6 per 100,000 person-years
- Possible explanations:
- Genetic founder effects
- High rates of specific genetic mutations
- Geographic isolation contributing to genetic homogeneity
Finnish Population
- High incidence: Up to 3.6 per 100,000 person-years
- Notable features:
- High C9orf72 repeat expansion frequency
- Genetic founder effect likely
- Comprehensive healthcare system with high detection rates
U.S. Military Veterans
- Increased risk: 1.5-2 times higher than general population
- Specific cohorts:
- Gulf War veterans show particularly elevated risk
- Vietnam-era veterans also show higher rates
- Ongoing research into specific exposures and environmental factors
Mortality and Survival Rates
Global Mortality Trends
- Annual ALS deaths worldwide: Approximately 80,000-100,000
- Mortality rates: Generally parallel to incidence rates
- 1.1-2.6 per 100,000 person-years in Europe and North America
- 0.3-1.0 per 100,000 person-years in Asia and Africa
- Temporal trends:
- Slight increase in age-adjusted mortality in many regions
- Likely reflects improved diagnosis rather than true incidence changes
- Death certificate accuracy:
- Estimated 85-90% sensitivity for ALS as cause of death in developed countries
- Much lower in regions with limited neurological care access
Survival Statistics
- Median survival from symptom onset:
- Global average: 2-4 years
- 20-30% survive 5+ years
- 5-10% survive 10+ years
- <5% survive 20+ years
- Regional variations in survival:
- Longer median survival in developed regions (3-4 years)
- Shorter median survival in developing regions (2-3 years)
- Europe and North America: 3-5 years median
- Asia: 2-4 years median
- Africa and parts of Latin America: 1.5-3 years median
- Prognostic factors affecting survival:
- Age at onset (younger = longer survival)
- Site of onset (limb > bulbar > respiratory)
- Rate of progression in first 6 months
- Respiratory function at diagnosis
- Cognitive status
- Access to multidisciplinary care
Specific Country Survival Data
- United States:
- Median survival: 2.5-3.5 years
- 5-year survival rate: 20-25%
- Geographic and racial disparities in survival
- United Kingdom:
- Median survival: 2.5-4 years
- Improved survival trends over past two decades
- Japan:
- Median survival: 3-5 years
- Higher rates of ventilatory support affecting statistics
- Italy:
- Median survival: 3-4 years
- North-south gradient in survival outcomes
Healthcare and Economic Impact
Economic Burden by Region
- United States:
- Annual cost per patient: $50,000-$120,000
- Total annual economic burden: $1-2 billion
- Direct medical costs represent 50-60% of total
- Europe:
- Annual cost per patient: €40,000-€80,000
- Considerable variation between countries
- Higher costs in Scandinavian countries
- Asia:
- Limited comprehensive data
- Estimated annual costs $20,000-$40,000 per patient in developed Asian economies
- Lower reported costs in developing regions, often reflecting limited care access
- Global perspective:
- Total global economic burden estimated at $8-10 billion annually
- Indirect costs (lost income, caregiver time) often exceed direct medical costs
Healthcare System Factors
- Specialized care access:
- Multidisciplinary ALS clinics available in most developed countries
- Limited or no specialized care in many developing regions
- Geographic disparities even within developed countries
- Ventilation practices:
- Japan: 30-40% of patients receive tracheostomy ventilation
- United States and Europe: 1-5% receive tracheostomy ventilation
- Significant impact on survival statistics and costs
- Riluzole access:
- Near-universal coverage in Western Europe
- Variable coverage in Eastern Europe and Asia
- Limited access in Africa and parts of Latin America
- Regional care delivery models:
- Centralized specialist centers in UK, Netherlands
- Distributed network model in U.S., Germany
- Primarily general neurology care in many developing countries
Genetic Epidemiology
Familial ALS Frequency
- Global average: 5-10% of ALS cases report a family history
- Regional variations:
- Higher rates (8-10%) in European and North American populations
- Lower reported rates (3-5%) in Asian populations
- Limited data from Africa and South America
- Underestimation factors:
- Small family sizes
- Incomplete penetrance
- Early death from other causes
- Inadequate family history collection
- True rate likely higher than reported
Gene Mutation Frequency by Region
- C9orf72 repeat expansion:
- Highest frequency in Finland and Northern Europe (40-50% of familial ALS)
- Intermediate in North America and Western Europe (25-40% of familial ALS)
- Rare in East Asia (<5% of familial ALS)
- Virtually absent in some populations (e.g., indigenous African populations)
- SOD1 mutations:
- Relatively consistent worldwide (15-20% of familial ALS)
- A4V mutation common in North America (50% of SOD1 cases)
- D90A mutation prevalent in Finland and Sweden
- Higher overall SOD1 frequency in Asian populations compared to C9orf72
- FUS mutations:
- Higher frequency in Asian populations (5-10% of familial ALS)
- Lower in European populations (3-5% of familial ALS)
- Associated with younger onset
- TARDBP mutations:
- Relatively consistent worldwide (3-5% of familial ALS)
- Specific mutations show regional clustering
Sporadic ALS Genetic Factors
- C9orf72 in apparently sporadic cases:
- Present in 5-10% of sporadic ALS in European populations
- 1-3% in East Asian populations
- Suggests incomplete penetrance or inadequate family history
- Overall genetic component:
- Heritability estimates 40-60% even in apparently sporadic cases
- Polygenic risk scores showing promise in risk stratification
- Gene-environment interactions likely critical
Ethnic and Racial Variations
Racial Differences in the United States
- Incidence by race:
- Highest in non-Hispanic whites (2.0-2.8 per 100,000 person-years)
- Intermediate in Hispanic populations (1.2-1.8 per 100,000 person-years)
- Lower in African Americans (0.8-1.4 per 100,000 person-years)
- Lowest in Asian Americans (0.7-1.2 per 100,000 person-years)
- Age at onset differences:
- Earlier average onset in African Americans (56.3 years vs. 61.7 years in whites)
- Shorter diagnostic delay in whites compared to minorities
- Survival disparities:
- Shorter survival in African Americans (16 months vs. 19 months in whites)
- Likely influenced by healthcare access, socioeconomic factors, and biology
Global Indigenous Populations
- Native American populations: Lower reported rates compared to European-ancestry Americans
- Australian Aboriginal people: Limited data, but suggested lower rates than European Australians
- Māori people: Lower rates compared to New Zealanders of European descent
- Sami people (Northern Scandinavia): Rates similar to surrounding populations
- Interpretation challenges: Healthcare access versus true biological differences
Time Trends and Future Projections
Historical Trends
- 20th century patterns:
- Rising incidence in first half of 20th century, likely due to improved diagnosis
- Relatively stable rates from 1970s-1990s in most regions
- Recent decades (1990-present):
- Slight increase in age-adjusted incidence in many regions
- More pronounced in those over 75 years
- Likely reflects improved diagnosis in elderly and population aging
- Some evidence for true increases in specific regions
Future Projections
- Demographic impact:
- Global cases expected to increase by 69% by 2040 due to population aging
- Largest increases projected in developing countries with rapidly aging populations
- Regional projections:
- East Asia: 81% increase in ALS cases by 2040
- Africa: 42% increase projected
- Europe: 34% increase projected
- Healthcare system implications:
- Growing need for specialized ALS centers
- Increasing economic burden
- Resource allocation challenges in developing regions
Understanding the global and regional epidemiology of ALS is essential for healthcare planning, research prioritization, and identifying potential environmental and genetic risk factors. The observed geographical and ethnic variations suggest a complex interplay of genetic susceptibility and environmental influences that continues to be a focus of intensive research.
11. Recent Research & Future Prospects
The field of ALS research has experienced remarkable acceleration in recent years, with advances in understanding disease mechanisms, developing biomarkers, and testing novel therapeutic approaches. These developments offer hope for transforming the treatment landscape for ALS in the coming years.
Latest Research Breakthroughs
Genetic Discoveries
- Expanded genetic landscape:
- Over 30 genes now associated with ALS
- Recent discoveries include KIF5A, TBK1, TUBA4A, and CCNF
- Genetic factors identified in 70-80% of familial and 15-20% of sporadic ALS
- Genetic modifiers:
- UNC13A and ATXN2 variants affect disease progression
- Identification of protective genetic variants (e.g., specific EPHA4 variants)
- Polygenic risk scores development for sporadic ALS
- Genetic pleiotropy:
- Stronger recognition of ALS-FTD genetic overlap
- Links to other neurodegenerative diseases (e.g., progressive supranuclear palsy)
- Connections to immune-mediated disorders
Pathophysiological Insights
- RNA processing dysfunction:
- Nuclear-cytoplasmic transport defects
- Stress granule dynamics and liquid-liquid phase separation
- Aberrant RNA splicing patterns
- Cellular autonomy understanding:
- Recognition of oligodendrocyte involvement
- Microglial activation patterns
- Astrocyte neurotoxic signatures
- Protein propagation models:
- Evidence for prion-like spread of misfolded proteins
- Trans-synaptic transmission of TDP-43 pathology
- Implications for therapeutic targeting
- Metabolic alterations:
- Hypermetabolism mechanisms
- Mitochondrial dynamics disruption
- Alterations in lipid metabolism
- Vascular contributions:
- Blood-spinal cord barrier dysfunction
- Neurovascular unit alterations
- Angiogenic factor dysregulation
Biomarker Development
- Fluid biomarkers:
- Neurofilament light chain (NfL): Validated in blood and CSF for diagnosis and prognosis
- Phosphorylated neurofilament heavy chain (pNfH): Correlates with disease progression
- TDP-43 detection methods in biofluids
- MicroRNA panels with high sensitivity/specificity
- Inflammatory marker profiles
- Neuroimaging advances:
- Multimodal MRI approaches identifying presymptomatic changes
- PET ligands for neuroinflammation visualization
- Quantitative muscle MRI as disease progression marker
- Advanced diffusion tensor imaging metrics
- Electrophysiological markers:
- Electrical impedance myography (EIM)
- Motor unit number estimation (MUNE)
- Threshold tracking for axonal excitability
- Digital biomarkers:
- Smartphone-based assessment tools
- Speech analysis for bulbar function
- Accelerometer-based movement analysis
- Home-based vital capacity monitoring
Treatment Advances
- FDA approvals:
- Tofersen (Qalsody): First antisense oligonucleotide for SOD1 ALS (2023)
- Relyvrio (sodium phenylbutyrate/taurursodiol): Approved in 2022
- Target engagement demonstration:
- Proof-of-concept for genetic therapies
- Measurable biological effects in phase 1/2 trials
- Improved trial design with pharmacodynamic endpoints
- Platform trial development:
- HEALEY ALS Platform Trial: Testing multiple drugs simultaneously
- European TRICALS initiative
- Adaptive trial designs with interim analyses
- Advanced delivery systems:
- Improved blood-brain barrier penetration
- Intrathecal delivery optimization
- AAV vector improvements for gene therapy
Ongoing Research Areas
Gene-Targeted Therapies
- Antisense oligonucleotides (ASOs):
- Tofersen for SOD1-ALS: First approved ASO therapy
- C9orf72-targeting ASOs in phase 3 trials
- ATXN2-targeting ASOs to modify TDP-43 toxicity
- Gene therapy approaches:
- AAV-delivered microRNAs targeting SOD1
- Gene replacement strategies
- CRISPR/Cas9 approaches in preclinical development
- RNA-based therapeutics:
- Small interfering RNAs (siRNAs)
- MicroRNA modulation
- RNA editing technologies
Cellular Mechanisms and Targets
- Protein homeostasis modulation:
- Enhancing autophagy
- Proteasome activation
- Heat shock protein inducers
- Mitochondrial therapeutics:
- Mitochondrial biogenesis enhancers
- Oxidative stress reducers
- Mitochondrial transport facilitators
- Neuroinflammation targeting:
- Microglial modulation approaches
- NF-κB pathway inhibitors
- Complement cascade interference
- Metabolic approaches:
- Addressing hypermetabolism
- Ketogenic strategies
- Insulin pathway modulation
Stem Cell Research
- Neural stem cell approaches:
- Focus on glial replacement and trophic support
- Phase 2 trials showing mixed results
- Route of administration optimization
- Mesenchymal stem cells:
- Immunomodulatory mechanisms
- Trial results with modest benefits
- Autologous versus allogeneic sources
- Induced pluripotent stem cells (iPSCs):
- Patient-derived disease modeling
- Drug screening platforms
- Potential therapeutic applications
- Organoid development:
- Motor cortex organoids
- Spinal cord organoids
- Applications in drug screening
Technological Innovations
- Brain-computer interfaces:
- SWITCH system for computer control
- Implantable electrode arrays
- Non-invasive BCI development
- Assistive technology:
- Eye-tracking advancements
- Thought-to-speech devices
- Wearable robotics
- Respiratory support innovations:
- Diaphragm pacing optimization
- Novel non-invasive ventilation interfaces
- Portable ventilation technologies
- AI applications:
- Diagnostic algorithm development
- Predictive progression models
- Treatment response prediction
Future Research Directions
Precision Medicine Implementation
- Genetic stratification:
- Treatment selection based on genetic subtype
- Custom genetic therapy development
- Combination approaches for complex genetic backgrounds
- Biomarker-guided therapy:
- Treatment initiation timing optimized by biomarkers
- Real-time monitoring of treatment response
- Adaptive dosing based on biomarker changes
- Phenotypic classification:
- Beyond traditional onset classification
- Molecular subtypes identification
- Machine learning for cluster identification
Preventive Approaches
- Pre-symptomatic intervention:
- Genetic carrier screening and treatment
- PREVENT-ALS trial concept for SOD1 carriers
- Biomarker-triggered intervention
- Risk modification strategies:
- Lifestyle interventions for at-risk individuals
- Environmental exposure mitigation
- Anti-inflammatory preventive approaches
- Early disease modification:
- Ultra-early diagnosis initiatives
- Treatment at first symptoms
- Combined biomarker approaches for screening
Therapeutic Pipeline
- Promising therapeutics in development:
- Masitinib: Tyrosine kinase inhibitor in phase 3
- Pridopidine: Sigma-1 receptor agonist in phase 3
- CNM-Au8: Cellular energy metabolizer in phase 2/3
- Verdiperstat: Myeloperoxidase inhibitor in phase 2/3
- Ibudilast: PDE4 inhibitor with anti-neuroinflammatory properties in phase 2/3
- Novel trial designs:
- HEALEY ALS Platform Trial expansion
- Seamless phase 2/3 designs
- Basket trials for genetic subtypes
- Real-world evidence integration
- Combination therapy approaches:
- Targeting multiple pathogenic mechanisms simultaneously
- Genetic plus small molecule combinations
- Symptom management plus disease modification
Global Research Initiatives
- Project MinE:
- Whole genome sequencing of 15,000+ ALS patients
- Identification of novel genetic risk factors
- Data sharing and open science approach
- Answer ALS:
- Comprehensive multi-omic profiling
- iPSC lines from 1,000 patients
- Machine learning applications
- Clinical Trial Networks:
- NEALS (Northeast ALS Consortium)
- TRICALS (Treatment Research Initiative to Cure ALS)
- Australian MND Clinical Trials Network
- Global collaborative trial infrastructure
Potential Future Breakthroughs
Near-Term Prospects (1-5 years)
- Expanded genetic therapy options:
- Additional ASOs for specific mutations
- First gene replacement therapies
- Broader access to genetic testing
- Combination therapy protocols:
- Multiple approved drugs used in rational combinations
- Improved functional outcomes
- Personalized regimens based on disease characteristics
- Enhanced prognostic tools:
- Accurate survival prediction algorithms
- Treatment response prediction
- Trial eligibility optimization
Medium-Term Prospects (5-10 years)
- Disease subtyping transformation:
- Molecular classification replacing phenotypic classification
- Customized treatment pathways
- Multi-biomarker diagnostic approaches
- Regenerative approaches:
- Cell replacement strategies reaching clinical application
- Combined cell therapy and molecular approaches
- Targeted restoration of specific neural circuits
- Presymptomatic treatment paradigms:
- Regular screening of at-risk individuals
- Biomarker-triggered intervention protocols
- Prevention trials showing success in familial ALS
Long-Term Prospects (10+ years)
- Disease-halting therapies:
- Complete stabilization for specific genetic subtypes
- Early intervention preventing progression
- Combination protocols effective across ALS subtypes
- Functional restoration approaches:
- Reversal of established motor deficits
- Combined molecular and cellular therapies
- Neural circuit reestablishment
- Cure pathways:
- Definition of “cure” in ALS context
- Elimination of disease-causing mechanisms
- Prevention strategies for at-risk populations
The research landscape for ALS has never been more promising, with unprecedented momentum in understanding disease mechanisms and developing novel therapeutics. While significant challenges remain, particularly in addressing the heterogeneity of the disease and delivering therapies effectively to the central nervous system, the convergence of genetic insights, technology advancements, and collaborative research networks provides realistic hope for transforming ALS from a uniformly fatal disease to a manageable chronic condition in the foreseeable future.
12. Interesting Facts & Lesser-Known Insights
Beyond the clinical and scientific aspects of ALS lie fascinating historical elements, cultural connections, and lesser-known facts that provide a broader perspective on this complex disease.
Historical Perspectives
Famous Figures with ALS
- Lou Gehrig (1903-1941):
- New York Yankees baseball player whose diagnosis brought ALS to public attention
- Played 2,130 consecutive games earning the nickname “The Iron Horse”
- Gave the famous “luckiest man on the face of the earth” speech at Yankee Stadium
- Died just two years after diagnosis at age 37
- Stephen Hawking (1942-2018):
- Renowned theoretical physicist who lived with ALS for 55 years
- Diagnosed at age 21 with expected survival of 2 years
- Became one of the longest-surviving patients in history
- Continued groundbreaking scientific work despite complete paralysis
- Mao Zedong (1893-1976):
- Chinese revolutionary leader believed to have had ALS
- Symptoms began in the early 1970s
- Bilateral hand weakness progressing to generalized weakness
- Diagnosis remains subject to historical debate
- David Niven (1910-1983):
- British actor and novelist
- Initially misdiagnosed before ALS confirmation
- Continued working even as his voice deteriorated
- Morrie Schwartz (1916-1995):
- Sociology professor whose battle with ALS was chronicled in “Tuesdays with Morrie”
- Book sold over 14 million copies and raised significant awareness
Historical Understanding Evolution
- Pre-Charcot observations:
- Ancient Egyptian medical papyri contain descriptions consistent with motor neuron disease
- 18th-century medical texts described progressive paralytic conditions without clear etiology
- Early terminology confusion:
- Initially classified among “progressive muscular atrophies”
- Multiple eponymous terms used in different countries
- Diagnostic criteria unstandardized until mid-20th century
- World War II impact:
- Accelerated neurological research and understanding
- Veterans’ studies contributed to epidemiological insights
- Military research funding advanced understanding
- The “ALS Bucket Challenge” phenomenon (2014):
- Raised over $115 million for ALS research
- Involved 17 million people uploading videos
- Led to significant research acceleration
- Funded important genetic discoveries
Unusual Scientific Facts
Exceptional Cases and Peculiarities
- Ultra-long survivors:
- Approximately 1 in 1,000 patients survive 20+ years
- Statistical outliers provide research opportunities
- Protective genetic modifiers likely present
- Reversible ALS mimics:
- Lead poisoning can present nearly identically to ALS
- Multifocal motor neuropathy may initially resemble ALS but responds to treatment
- Hyperparathyroidism occasionally presents with ALS-like symptoms
- Geographical clusters:
- Western Pacific ALS-parkinsonism-dementia complex
- French Alpine valley clusters
- Western New Guinea high-incidence regions
- Potential environmental triggers being investigated
- Cognitive preservation paradox:
- Intact cognition despite profound physical disability
- Preservation of eye movements even in complete paralysis
- Sensory systems typically unaffected
- Extraocular muscle sparing:
- Eye muscles preserved even in end-stage disease
- Multiple theories including unique innervation patterns
- Potential biological protection mechanisms of interest for therapy
Biological Curiosities
- Exercise paradox:
- Fitness and athleticism associated with higher risk
- Yet exercise therapy beneficial after diagnosis
- Possibly explained by “what’s good for the brain may stress motor neurons”
- Hypermetabolism:
- Patients burn 300-500 more calories daily than expected
- Present even before diagnosis in many cases
- Correlates with faster progression in some studies
- Military service connection:
- 1.5-2 times higher risk in veterans
- Particularly strong in Gulf War veterans
- Multiple potential factors including exposures, physical demands, and stress
- Cancer inverse relationship:
- Lower cancer rates in ALS patients
- Possibly related to shared p53 pathway differences
- Potential therapeutic implications in development
- Left-handedness association:
- Modest but consistent association with ALS risk
- May reflect developmental or neuroanatomical factors
- Links to cerebral dominance patterns
Myths and Misconceptions
Common Misconceptions
- “ALS affects only older people”:
- While median age is 55-65, young-onset cases occur
- 10% of cases begin before age 45
- Juvenile ALS (before age 25) represents about 1% of cases
- “ALS always progresses rapidly”:
- 10-20% have slow progression
- “Benign” variants can have decade-plus survival
- Progression rate highly variable between individuals
- “ALS is primarily a hereditary disease”:
- Only 5-10% report clear family history
- Though genetic factors contribute to many “sporadic” cases
- Most cases appear without family history
- “ALS affects only motor function”:
- 50% have some degree of cognitive/behavioral changes
- 15% develop frontotemporal dementia
- Non-motor systems increasingly recognized as involved
- “All ALS is the same disease”:
- Growing recognition of ALS as a syndrome rather than single disease
- Multiple subtypes with distinct biology
- Treatment responses vary significantly between patients
Debunked Theories
- Mercury dental amalgam link:
- Extensively studied and no association found
- Controlled studies show no risk difference
- Vaccination triggering:
- No evidence supporting vaccine-induced ALS
- Population studies show no temporal relationship with vaccination
- Electric shock as primary cause:
- Earlier theories suggested electrical injury causation
- Modern studies show minimal or inconsistent association
- “ALS is contagious”:
- No evidence for person-to-person transmission
- Prion-like mechanisms refer to protein spread within an individual, not between people
- “ALS patients lose all communication ability”:
- Modern assistive technology enables communication throughout disease
- Eye-tracking and brain-computer interfaces preserve communication ability
Cultural and Social Dimensions
Literary and Film Representations
- “Tuesdays with Morrie” (1997):
- Mitch Albom’s memoir about his professor’s ALS journey
- One of the most influential modern works about the disease
- Widely assigned in medical humanities courses
- “The Theory of Everything” (2014):
- Biographical film about Stephen Hawking
- Eddie Redmayne won the Academy Award for his portrayal
- Criticized by some for romanticizing disability
- “You’re Not You” (2014):
- Film starring Hilary Swank as an ALS patient
- Explored caregiver relationships and end-of-life choices
- Mixed reception from ALS community
- “The Pride of the Yankees” (1942):
- Early film portrayal of Lou Gehrig’s story
- Gary Cooper’s iconic performance
- Notable for its era’s limited understanding of ALS
Support Communities and Advocacy
- Patient-led innovations:
- ALS patients have developed numerous adaptive technologies
- Eric Valor developed eye-controlled home automation while paralyzed
- Steve Gleason (former NFL player) pioneered accessibility technologies
- ALS Association impact:
- Founded in 1985, now the largest ALS-specific organization
- Funds research, advocacy, and patient services
- Administers a network of certified treatment centers
- Team Gleason initiatives:
- Founded by former NFL player Steve Gleason
- Focus on technology access and independence
- Successfully advocated for the Steve Gleason Act to improve Medicare coverage
- Project MinE:
- Patient-initiated genetic research project
- Crowdfunded genome sequencing
- Discovered several ALS genes through this unique approach
Professional Impact
- Medical profession involvement:
- Higher rates among physicians in some studies
- Notable cases including neurologists who studied their own disease
- Dr. Richard K. Olney: ALS researcher who developed ALS and studied himself
- Professional athletes:
- NFL players have 4x higher mortality from ALS
- Professional Italian soccer players show 6x elevated risk
- Numerous high-profile cases raising awareness (Steve Gleason, Tim Green, Dwight Clark)
- Military legacy:
- Veteran-focused research initiatives
- Special VA programs for ALS patients
- Presumptive service connection for all veterans with ALS regardless of when or where they served
Practical Insights
Communication Approaches
- Voice banking technology:
- Patients can record their voice before speech deterioration
- Synthetic speech created from recordings
- Preserves vocal identity for communication devices
- Eye-tracking evolution:
- Modern systems allow typing 15-20 words per minute
- Calibration accommodates progressive changes
- Integration with smart home systems
- Brain-computer interfaces:
- Implantable arrays allow direct neural control
- Non-invasive options in development
- Some patients achieve near-typical typing speeds
Quality of Life Factors
- Psychological well-being paradox:
- Studies show psychological well-being often maintained despite physical decline
- “Well-being gap” between patient self-reports and observer assumptions
- Adaptation and shifting values contribute to resilience
- Employment adaptations:
- Stephen Hawking continued academic work throughout his illness
- Remote work technologies enabling extended careers
- Modified work environments allowing continued productivity
- Relationship dynamics:
- Impact on intimacy and partners
- Changing roles within families
- Caregiver-patient relationship evolution
Lesser-Known Symptoms
- Emotional lability:
- Uncontrollable laughing or crying disproportionate to emotions
- Present in 50% of patients at some point
- Often mistaken for depression or inappropriate reactions
- Metabolic changes:
- Weight loss despite adequate caloric intake
- Altered lipid metabolism
- Changes in taste and smell in some patients
- Sleep architecture disruption:
- REM sleep abnormalities
- Fragmented sleep patterns
- Sleep-disordered breathing preceding respiratory symptoms
Future Possibilities
Technological Frontiers
- Direct brain communication:
- Stentrode™ endovascular electrode array
- Allows thought-to-text without open brain surgery
- First implants showing promising results
- Virtual reality applications:
- Immersive experiences despite physical limitations
- Therapeutic applications for psychological well-being
- VR-based physical therapy showing early promise
- Robotic exoskeletons:
- External support systems enabling movement
- Currently in early stages for ALS applications
- Potential for maintained mobility and independence
Social and Care Innovations
- Housing design evolution:
- Smart homes specifically designed for ALS needs
- Community living models with shared care resources
- Architectural innovations improving accessibility
- Remote care models:
- Telemedicine approaches reducing travel burden
- Home monitoring systems with predictive analytics
- Virtual multidisciplinary clinics
- Financial models:
- Insurance innovations for catastrophic disease costs
- Long-term care financing mechanisms
- Advocacy for policy changes to address financial toxicity
ALS continues to intrigue, challenge, and inspire both the scientific community and the broader public. These lesser-known aspects provide important context to the disease beyond its clinical manifestations, highlighting the human experience, historical significance, and cultural impact of this complex condition. As research advances and awareness grows, many of these interesting elements will continue to evolve, potentially leading to new insights that further our understanding of this devastating yet fascinating disorder.