
⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
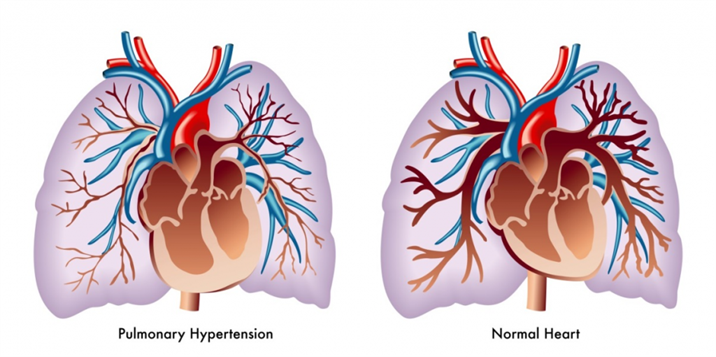
Comprehensive Report on Pulmonary Hypertension
1. Overview
What is Pulmonary Hypertension?
Pulmonary hypertension (PH) is a complex, progressive cardiovascular condition characterized by abnormally elevated blood pressure in the pulmonary arterial circulation. While normal mean pulmonary arterial pressure (mPAP) at rest is approximately 14 mmHg, pulmonary hypertension is clinically defined as a mPAP ≥20 mmHg at rest as measured by right heart catheterization.
This elevation in pressure forces the right ventricle to work harder to pump blood through the lungs, leading to right ventricular strain, hypertrophy, and eventually failure if left untreated. Pulmonary hypertension is not a single disease but rather a hemodynamic abnormality common to a diverse array of clinical conditions.
Classification
The World Health Organization (WHO) classifies pulmonary hypertension into five distinct groups based on etiology, pathophysiology, and therapeutic approaches:
Group 1: Pulmonary Arterial Hypertension (PAH)
- Idiopathic PAH
- Heritable PAH
- Drug and toxin-induced PAH
- PAH associated with connective tissue disease, HIV infection, portal hypertension, congenital heart disease, schistosomiasis
Group 2: Pulmonary Hypertension due to Left Heart Disease
- Left ventricular systolic/diastolic dysfunction
- Valvular disease
- Congenital/acquired left heart inflow/outflow tract obstruction
Group 3: Pulmonary Hypertension due to Lung Diseases and/or Hypoxia
- Chronic obstructive pulmonary disease (COPD)
- Interstitial lung disease
- Sleep-disordered breathing
- Chronic exposure to high altitude
Group 4: Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
- Pulmonary hypertension resulting from blood clots in the lungs
Group 5: Pulmonary Hypertension with Unclear Multifactorial Mechanisms
- Hematological disorders
- Systemic disorders (sarcoidosis, vasculitis)
- Metabolic disorders
- Complex congenital heart disease
Affected Body Parts/Organs
Pulmonary hypertension primarily affects:
- Pulmonary Vasculature: Increased pressure and vascular remodeling in pulmonary arteries and arterioles
- Right Ventricle: Hypertrophy, dilation, and eventually failure due to increased workload
- Lungs: Impaired gas exchange and ventilation-perfusion mismatch
- Systemic Circulation: Reduced cardiac output affecting all organs
- Liver and Kidneys: Secondary congestion due to right heart failure
Prevalence and Significance
Pulmonary hypertension affects people of all ages, races, and ethnic backgrounds, though specific forms have different demographic patterns.
Prevalence:
- Group 1 (PAH): 15-60 cases per million population
- IPAH (a subset of Group 1): 5-10 cases per million
- Group 2 (left heart disease): Most common form, present in up to 80% of patients with severe heart failure
- Group 3 (lung disease): Present in 30-70% of patients with advanced COPD
- Group 4 (CTEPH): 3-30 cases per million
- Overall: Estimated 1% of the global population may have some form of pulmonary hypertension
Significance:
- High morbidity and mortality if untreated
- Substantial impact on quality of life and functional capacity
- Economic burden through hospitalization, specialized medications, and lost productivity
- Without treatment, median survival for PAH is approximately 2.8 years from diagnosis
- Major advances in treatment have improved survival, but many forms remain incurable
- Healthcare costs for PAH patients average $50,000-$100,000 annually in developed countries
2. History & Discoveries
First Identification
The first clinical descriptions of pulmonary hypertension date back to the late 19th and early 20th centuries:
- In 1865, German physician Julius Klob described thickening of the pulmonary arteries in a patient at autopsy but did not connect it to clinical symptoms
- In 1891, Ernst von Romberg described “sclerosis of the pulmonary arteries” but also didn’t associate it with clinical manifestations
- In 1901, Dr. Abel Ayerza from Argentina described a syndrome of pulmonary arterial hypertension with cyanosis, which became known as “Ayerza’s disease” or “black cardiacs”
However, the first comprehensive clinical description and linkage to pathophysiology is attributed to Dr. André Cournand and Dr. Dickinson Richards, who in the 1940s pioneered cardiac catheterization techniques that allowed measurement of pressures within the heart and pulmonary circulation.
Major Discoveries and Breakthroughs
1940s-1950s: Diagnostic Advancements
- Development of cardiac catheterization techniques by Cournand and Richards (shared 1956 Nobel Prize in Medicine)
- First hemodynamic characterization of pulmonary hypertension
1960s-1970s: Epidemiological Insights
- 1967-1968: Aminorex (appetite suppressant) epidemic in Europe leads to recognition of drug-induced PAH
- 1973: WHO holds first meeting on pulmonary hypertension, establishing initial classification
1980s: Pathophysiological Understanding
- 1980s: Discovery of endothelin, nitric oxide, and prostacyclin pathways in vascular regulation
- 1981: First heart-lung transplant performed for pulmonary hypertension
1990s: Treatment Breakthroughs
- 1995: FDA approval of epoprostenol (Flolan), the first specific treatment for PAH
- 1998: Identification of BMPR2 gene mutations in familial PAH
2000s: Expanded Treatment Options
- 2001: Bosentan approved as first oral therapy for PAH
- 2002: Updated clinical classification system (Venice classification)
- 2005-2009: Multiple new drug approvals (sildenafil, tadalafil, ambrisentan)
2010s: Refined Understanding and Management
- 2013: Updated diagnostic definition and classification (Nice classification)
- 2013: Riociguat approved for PAH and inoperable CTEPH
- 2015: Evidence for upfront combination therapy
- 2015-2019: Selexipag, macitentan and other new drugs approved
2020s: Precision Medicine Approaches
- Genetic testing becoming standard in PAH evaluation
- Biomarker-driven treatment strategies
- Increasing focus on right ventricular function
- Novel therapeutic targets including inflammation and metabolism
Evolution of Medical Understanding
The conceptualization of pulmonary hypertension has evolved dramatically over the past century:
Early Understanding (pre-1950s):
- Primarily a pathological curiosity identified at autopsy
- Limited understanding of underlying mechanisms
- No effective treatments available
- Often misdiagnosed as anxiety or deconditioning
Middle Era (1950s-1980s):
- Recognition as a distinct hemodynamic condition
- Understanding of basic pathophysiology
- Limited treatment options primarily addressing symptoms
- Classification based on anatomical changes
Modern Era (1990s-2010s):
- Recognition of multiple pathogenic pathways
- Development of targeted therapies
- Establishment of comprehensive classification systems
- Improved diagnostic capabilities
- Recognition of genetic contributions
Current Understanding (2010s-Present):
- Pulmonary hypertension viewed as a complex, multifactorial syndrome
- Recognition of the importance of early diagnosis and treatment
- Emphasis on multidisciplinary care at specialized centers
- Personalized treatment approaches based on patient characteristics
- Understanding of molecular and genetic underpinnings
- Recognition of right ventricular function as a key determinant of outcomes
This evolution reflects a transition from a uniformly fatal condition with no treatments to a chronic, manageable disease with multiple therapeutic options and improved survival for many patients.
3. Symptoms
Early Symptoms
Early symptoms of pulmonary hypertension are often subtle and non-specific, frequently leading to delayed diagnosis. These typically develop gradually and may be attributed to deconditioning, aging, or other common conditions:
Most Common Early Symptoms:
- Dyspnea (shortness of breath) with exertion: Initially occurs only with significant activity
- Fatigue and weakness: Often described as unexplained or out of proportion to activity level
- Dizziness or lightheadedness: Especially during physical activity
- Non-productive cough: Typically dry and persistent
Less Common Early Symptoms:
- Chest discomfort: Often atypical, described as pressure rather than pain
- Palpitations: Awareness of heartbeat, especially during activity
- Early satiety: Feeling full quickly when eating
- Mild lower extremity edema: Subtle swelling of the ankles or feet
These early symptoms are often overlooked or misattributed to aging, obesity, deconditioning, asthma, or anxiety, contributing to the average 2-year delay in diagnosis from symptom onset.
Advanced-Stage Symptoms
As pulmonary hypertension progresses, symptoms become more pronounced and significantly impact quality of life:
Cardiopulmonary Symptoms:
- Severe dyspnea: Occurs with minimal exertion or even at rest
- Syncope (fainting): Due to inadequate cerebral perfusion, especially with exertion
- Hemoptysis: Coughing up blood due to rupture of pulmonary vessels
- Chest pain: More prominent and may mimic angina
- Palpitations: More frequent, may represent arrhythmias
Right Heart Failure Symptoms:
- Pronounced peripheral edema: Significant swelling of legs, ankles
- Ascites: Fluid accumulation in the abdomen
- Distended neck veins: Visible jugular venous distention
- Hepatomegaly: Enlarged, sometimes tender liver
- Cyanosis: Bluish discoloration of lips, nail beds due to poor oxygenation
Systemic Symptoms:
- Severe fatigue: Profound exhaustion limiting basic activities
- Weight loss: Cardiac cachexia in advanced disease
- Hoarseness: Due to compression of the left recurrent laryngeal nerve by enlarged pulmonary artery (Ortner’s syndrome)
- Raynaud’s phenomenon: In connective tissue disease-associated PAH
Common vs. Rare Symptoms
Common Symptoms (>50% of patients):
- Dyspnea on exertion
- Fatigue
- Chest discomfort/pain
- Dizziness/lightheadedness
- Lower extremity edema
- Palpitations
- Dry cough
Uncommon Symptoms (10-50% of patients):
- Syncope
- Hemoptysis
- Hoarseness
- Abdominal distention
- Anorexia and early satiety
- Right upper quadrant discomfort (liver congestion)
- Raynaud’s phenomenon (in certain types)
Rare Symptoms (<10% of patients):
- Angina-like pain at rest
- Orthopnea (shortness of breath when lying flat)
- Massive hemoptysis
- Pleuritic chest pain
- Clubbing of digits (more common in congenital heart disease-associated PAH)
- Symptoms of hyperviscosity (headache, visual disturbances) in Eisenmenger syndrome
Symptom Progression Over Time
The natural history of pulmonary hypertension symptoms typically follows a pattern, though the rate of progression varies considerably based on etiology, age, comorbidities, and treatment:
Symptom Progression Timeline:
Asymptomatic Phase:
- Pulmonary vascular changes may be occurring
- Normal or near-normal exercise capacity
- May last for years, especially in heritable forms
- Detectable only through screening of high-risk individuals
Early Symptomatic Phase (NYHA/WHO Functional Class II):
- Symptoms with moderate to significant exertion
- No symptoms at rest
- Subtle lifestyle modifications to accommodate limitations
- May last months to years
- Diagnosis often occurs in this phase
Intermediate Phase (NYHA/WHO Functional Class III):
- Symptoms with mild exertion
- Limited ordinary physical activity
- Comfortable only at rest
- Signs of right ventricular dysfunction may emerge
- Typical duration: months to a few years without treatment
Advanced Phase (NYHA/WHO Functional Class IV):
- Symptoms at rest or with minimal activity
- Inability to perform any physical activity without discomfort
- Signs of right heart failure prominent
- Without advanced therapy, typically lasts weeks to months
- High mortality risk
Factors Affecting Symptom Progression:
- Type of pulmonary hypertension (Group 1-5)
- Age at onset (younger patients often progress more slowly)
- Genetic factors (certain mutations associate with more rapid progression)
- Comorbidities
- Access to and response to therapy
- Development of complications
With modern therapies, this natural history can be significantly altered, with many patients experiencing stabilization or even improvement in symptoms, particularly in PAH (Group 1). However, the disease remains progressive for most patients over the long term, requiring ongoing monitoring and treatment adjustments.
4. Causes
Biological Causes
The pathophysiological mechanisms of pulmonary hypertension vary by classification group but generally involve:
Pathophysiological Mechanisms:
Vasoconstriction: Imbalance between vasodilators and vasoconstrictors
- Decreased production of vasodilators (nitric oxide, prostacyclin)
- Increased production of vasoconstrictors (endothelin-1, thromboxane A2)
Vascular Remodeling:
- Proliferation of smooth muscle cells
- Fibroblast activation and proliferation
- Endothelial cell dysfunction
- Extracellular matrix deposition
In-situ Thrombosis:
- Endothelial dysfunction promoting thrombosis
- Abnormal platelet function
- Decreased fibrinolysis
Inflammation:
- Perivascular inflammatory cell infiltration
- Increased cytokine production
- Autoantibody involvement in some forms
Metabolic Dysfunction:
- Mitochondrial abnormalities
- Warburg effect (shift to glycolytic metabolism)
- Altered fatty acid oxidation
Specific Biological Causes by Group:
Group 1 (PAH):
- BMPR2 mutations affecting BMP signaling pathway
- Mutations in other genes (ACVRL1, ENG, SMAD9, CAV1, KCNK3)
- Endothelial cell dysfunction
- Excessive vasoconstriction
- Uncontrolled proliferation of vascular cells
Group 2 (Left Heart Disease):
- Passive backward transmission of pressure from left heart
- Reactive pulmonary vasoconstriction
- Eventual vascular remodeling
Group 3 (Lung Disease/Hypoxia):
- Hypoxic pulmonary vasoconstriction
- Destruction of pulmonary vascular bed
- Inflammatory mechanisms from underlying lung disease
- Mechanical effects of hyperinflation
Group 4 (CTEPH):
- Mechanical obstruction of pulmonary vessels by organized thrombi
- Failure of thrombus resolution
- Secondary vascular remodeling in non-obstructed vessels
- Abnormal fibrinolysis
Group 5 (Multifactorial Mechanisms):
- Various mechanisms depending on underlying disorder
- Often involves multiple pathways
Environmental Causes
Several environmental factors have been implicated in the development of pulmonary hypertension:
Drugs and Toxins:
- Definite Association:
- Aminorex
- Fenfluramine/dexfenfluramine (diet pills)
- Toxic rapeseed oil
- Methamphetamines
- Dasatinib (cancer medication)
- Likely Association:
- Amphetamines
- L-tryptophan
- St. John’s wort
- Chemotherapy agents (mitomycin C, cyclophosphamide)
- Interferon alpha and beta
Other Environmental Factors:
- High Altitude: Chronic hypoxia leading to pulmonary vasoconstriction
- Air Pollution: Associated with worse outcomes in existing PH
- Organic Solvents: Possible association with PAH development
- Radiation Exposure: Radiation pneumonitis can lead to PH
- Mining Exposures: Certain mineral dusts associated with PH
Genetic and Hereditary Factors
Genetic factors play a significant role, particularly in Group 1 PAH:
Heritable PAH:
- 6-10% of PAH patients have family history
- Autosomal dominant inheritance with incomplete penetrance (20-30%)
- Female predominance suggesting hormonal modifiers
Key Genetic Mutations:
- BMPR2: Most common (80% of heritable PAH, 20% of idiopathic PAH)
- ALK1/ACVRL1, ENG: Associated with hereditary hemorrhagic telangiectasia
- CAV1: Caveolin-1 gene affecting endothelial function
- KCNK3: Potassium channel mutation
- TBX4: Associated with developmental abnormalities
- EIF2AK4: Biallelic mutations in pulmonary veno-occlusive disease
- SOX17: Transcription factor affecting vascular development
Genetic Risk Factors:
- Multiple rare variants with small individual effects
- Common variants identified through genome-wide association studies
- Genetic modifiers affecting penetrance
Known Triggers and Exposure Risks
Several factors are known to trigger or exacerbate pulmonary hypertension:
Infectious Triggers:
- HIV infection: 0.5% develop PAH
- Schistosomiasis: Leading global cause of PAH
- Hepatitis B and C (associated with portopulmonary hypertension)
Physiological Triggers:
- Pregnancy: Can unmask or worsen PAH
- Exercise: May exacerbate symptoms and reveal latent disease
- High altitude exposure: Acute or chronic effects
Systemic Disease Associations:
- Connective tissue diseases (scleroderma, SLE, mixed CTD)
- Liver disease with portal hypertension
- Congenital heart defects (especially unrestricted left-to-right shunts)
- Hematological disorders (chronic hemolytic anemias, myeloproliferative disorders)
Mechanical Factors:
- Pulmonary embolism (acute or chronic)
- Pulmonary vein stenosis
- Extrinsic compression of pulmonary vessels
Understanding these diverse causes is essential for proper classification, which guides treatment decisions and prognostication. The multifactorial nature of most forms of pulmonary hypertension highlights the need for comprehensive evaluation and often multidisciplinary management.
5. Risk Factors
Demographic Risk Factors
Age:
- PAH (Group 1) has a bimodal distribution with peaks in the 30s and 60s
- Group 2 PH increases with age, most common in 65+ years
- Group 3 PH typically affects older adults with lung disease
- CTEPH (Group 4) most common in 40-60 year range
- Overall prevalence increases with age across all groups
Gender:
- Group 1 PAH: Female predominance (F:M ratio 2-4:1)
- IPAH particularly affects women in childbearing years
- Group 2 PH: More balanced gender distribution
- Group 3 PH: Varies with underlying lung disease (COPD more common in men)
- CTEPH: Slight female predominance
- Male gender associated with worse outcomes in PAH despite lower prevalence
Race/Ethnicity:
- IPAH reported more frequently in Caucasians but likely underdiagnosed in minorities
- Sarcoidosis-associated PH more common in African Americans
- Schistosomiasis-PAH predominantly affects populations in endemic regions (Africa, South America, Asia)
- Possible genetic susceptibility variations between ethnic groups
- Healthcare disparities affect diagnosis rates across different populations
Geographic Factors:
- High-altitude residing populations: Higher risk of Group 3 PH
- Schistosomiasis-endemic regions: Higher rates of Group 1 PAH
- Coal mining regions: Higher rates of Group 3 PH
- Urban areas with high air pollution: Possible increased risk and worse outcomes
Environmental and Occupational Risk Factors
Occupational Exposures:
- Mining occupations: Exposure to coal, silica dust
- Agricultural workers: Exposure to pesticides and organic dusts
- Chemical industry workers: Exposure to solvents and toxic chemicals
- Healthcare workers: Potential exposure to chemotherapeutic agents
- Military service: Exposure to burn pits, chemical agents
Environmental Exposures:
- Altitude: Residence above 2500m increases risk
- Air pollution: Particulate matter and industrial emissions
- Indoor air pollution: Biomass fuel use in developing countries
- Asbestos: Associated with restrictive lung disease and secondary PH
- Radiation: Therapeutic radiation to chest may damage pulmonary vessels
Lifestyle Factors:
- Smoking: Major risk factor for COPD and associated Group 3 PH
- Intravenous drug use: Risk of HIV, hepatitis, and particulate embolization
- Anorexigen use: Appetite suppressants like fenfluramine
- Stimulant use: Amphetamines, cocaine, methamphetamine
- Physical activity: Extreme exercise may unmask susceptibility in predisposed individuals
Genetic Risk Factors
Heritable Factors:
- First-degree relatives of PAH patients have 10-fold increased risk
- BMPR2 mutation carriers have 20% (male) to 42% (female) lifetime risk of developing PAH
- Higher risk with multiple genetic mutations in related pathways
- Genetic anticipation observed in some families (earlier onset in subsequent generations)
- Epigenetic modifications may affect gene expression and disease manifestation
Genetic Syndromes Associated with PH:
- Down syndrome: Increased risk of PAH, particularly with congenital heart disease
- Hereditary hemorrhagic telangiectasia (HHT): ACVRL1 and ENG mutations
- Pulmonary veno-occlusive disease: EIF2AK4 mutations
- Congenital diaphragmatic hernia: TBX4 mutations
- Filamin A mutations: X-linked form of PAH
Impact of Pre-existing Conditions
Cardiovascular Conditions:
- Congenital heart disease: 5-10% develop PAH (Eisenmenger syndrome)
- Left heart failure: Up to 80% develop secondary PH
- Valvular heart disease: Mitral stenosis/regurgitation particularly significant
- Coronary artery disease: Can contribute to Group 2 PH
Pulmonary Conditions:
- COPD: 30-70% develop some degree of PH
- Interstitial lung disease: 30-60% develop PH
- Sleep apnea: 17-53% have PH, usually mild to moderate
- Pulmonary fibrosis: Strong association with PH, worse prognosis
- History of pulmonary embolism: 3-4% develop CTEPH
Systemic Conditions:
Connective tissue diseases:
- Systemic sclerosis: 8-12% develop PAH
- SLE: 1-5% develop PAH
- Mixed connective tissue disease: 10-45% develop PAH
- Rheumatoid arthritis: Increased risk of Group 3 PH
Liver disease:
- Portal hypertension: 2-6% develop portopulmonary hypertension
- Chronic hepatitis: Associated with PAH
Infectious diseases:
- HIV: 0.5% develop PAH
- Schistosomiasis: Leading global cause of PAH
Hematologic disorders:
- Chronic hemolytic anemias (sickle cell, thalassemia): Up to 10% develop PAH
- Myeloproliferative disorders: Can lead to Group 5 PH
- Splenectomy: Increases risk of CTEPH and Group 1 PAH
Metabolic disorders:
- Thyroid disorders (both hypo- and hyperthyroidism)
- Glycogen storage diseases
- Diabetes: May worsen outcomes in existing PH
Risk Factor Interactions: Many patients have multiple risk factors that interact synergistically:
- Female gender + BMPR2 mutation: Higher penetrance
- Connective tissue disease + male gender: Worse prognosis
- HIV + intravenous drug use: Higher PAH risk
- Multiple genetic variants: Gene-gene interactions
- Genetic predisposition + environmental triggers: Gene-environment interactions
Understanding these risk factors enables targeted screening of high-risk populations, potentially allowing earlier diagnosis and intervention before significant right ventricular dysfunction develops.
6. Complications
Direct Complications of Pulmonary Hypertension
Right Ventricular Dysfunction and Failure:
- Progressive right ventricular hypertrophy
- Right ventricular dilation and dysfunction
- Tricuspid regurgitation due to annular dilation
- Right ventricular ischemia from increased oxygen demand
- Right heart failure with systemic congestion
- Reduced cardiac output and cardiogenic shock in advanced stages
Arrhythmias:
- Atrial arrhythmias (15-25% of patients)
- Atrial fibrillation
- Atrial flutter
- Atrial tachycardia
- Ventricular arrhythmias (less common but more dangerous)
- Sudden cardiac death (6-13% of PAH patients)
Hemoptysis:
- Minor hemoptysis: Rupture of hypertrophied bronchial vessels
- Massive hemoptysis: Potentially life-threatening complication
- More common in CTEPH, Eisenmenger syndrome, and pulmonary veno-occlusive disease
Thrombotic Complications:
- In-situ pulmonary arterial thrombosis
- Paradoxical embolism through right-to-left shunting
- Deep vein thrombosis due to venous stasis
- Thrombotic microangiopathy in systemic sclerosis-associated PAH
Hypoxemic Complications:
- Ventilation-perfusion mismatching
- Right-to-left shunting through patent foramen ovale
- Reduced mixed venous oxygen saturation
- Secondary polycythemia from chronic hypoxemia
Hepatic and Renal Complications:
- Congestive hepatopathy from right heart failure
- Cardiorenal syndrome
- Hepatic dysfunction affecting drug metabolism
- Ascites and peripheral edema
Long-term Impact on Organs and Overall Health
Cardiovascular System:
- Progressive right ventricular failure
- Reduced exercise capacity
- Increased susceptibility to cardiovascular stressors
- Paradoxical left ventricular underfilling
- Coronary perfusion abnormalities
Pulmonary System:
- Progressive vascular remodeling
- Reduced pulmonary compliance
- Increased dead space ventilation
- Impaired gas exchange efficiency
- Increased work of breathing
Hepatic System:
- Chronic passive congestion
- Elevated liver enzymes
- Reduced synthetic function with advanced disease
- Fibrosis and “cardiac cirrhosis” in long-standing disease
Renal System:
- Prerenal azotemia from reduced cardiac output
- Venous congestion impairing renal function
- Diuretic resistance in advanced disease
- Progressive decline in GFR with worsening right heart failure
Hematologic System:
- Secondary erythrocytosis in hypoxemic patients
- Iron deficiency (30-63% of PAH patients)
- Thrombocytopenia in some patients (especially CTEPH)
- Acquired von Willebrand syndrome with severe PH
Musculoskeletal System:
- Skeletal muscle deconditioning
- Respiratory muscle fatigue
- Bone loss from inactivity and medications
- Joint pain and stiffness in connective tissue disease-associated PAH
Neurological System:
- Cognitive impairment from chronic hypoxemia
- Syncope and presyncope from reduced cerebral perfusion
- Anxiety and depression (prevalent in 20-40% of patients)
- Sleep disturbances
Endocrine and Metabolic:
- Right ventricular myocardial insulin resistance
- Altered adipokine profiles
- Metabolic shifts to glycolysis
- Catabolism and cachexia in advanced disease
Disability and Fatality Rates
Functional Disability:
- WHO/NYHA Functional Class distribution at diagnosis:
- Class I: 5-10%
- Class II: 25-35%
- Class III: 40-50%
- Class IV: 15-20%
- 57-75% of patients report reduced ability to perform daily activities
- 25-65% unable to work due to PH (varies by country and healthcare system)
50% report significant impairment in quality of life
- Progressive reduction in 6-minute walk distance correlates with worsening disability
Hospitalization Rates:
- Annual hospitalization rate: 30-50% of PAH patients
- Mean hospital stay: 5-9 days
- Readmission within 30 days: 20-25%
- ICU admission: 10-18% of hospitalized PH patients
Mortality Statistics:
Historical untreated median survival in IPAH: 2.8 years from diagnosis
Modern treated survival in PAH:
- 1-year survival: 85-93%
- 3-year survival: 68-80%
- 5-year survival: 55-75%
- 10-year survival: 30-45%
Mortality by WHO Group:
- Group 1 (PAH): 5-15% annual mortality
- Group 2 (left heart disease): Varies widely based on underlying cardiac condition
- Group 3 (lung disease): 5-year survival 30-50%
- Group 4 (CTEPH): 10-15% 3-year mortality if not surgical candidates; 90% 5-year survival after successful PEA
- Group 5: Highly variable depending on underlying cause
Mortality Risk Factors:
- WHO/NYHA Functional Class III-IV
- Male gender
- Advanced age (>65 years)
- Connective tissue disease etiology
- Renal dysfunction
- Right atrial pressure >15 mmHg
- Cardiac index <2 L/min/m²
- 6-minute walk distance <300 meters
- Brain natriuretic peptide >180 pg/mL
- Pericardial effusion on echocardiography
Despite advances in treatment, pulmonary hypertension remains a life-shortening condition with substantial impact on quality of life. Early diagnosis, prompt initiation of appropriate therapy, and management at specialized centers are associated with improved outcomes.
7. Diagnosis & Testing
Common Diagnostic Procedures
Clinical Evaluation:
- Comprehensive medical history
- Family history assessment
- Physical examination focusing on cardiopulmonary signs
- Functional capacity assessment using WHO/NYHA classification
- Evaluation for underlying causes and associated conditions
Diagnostic Algorithm: The diagnostic approach typically follows a stepwise process:
- Recognition of symptoms suggesting PH
- Detection of PH by echocardiography
- Identification of the most common causes (Groups 2 and 3)
- Differentiation between Group 1 (PAH) and Group 4 (CTEPH)
- Confirmation with right heart catheterization
- Characterization of specific PAH subtype (Group 1)
Risk Assessment: Several validated risk assessment tools are used:
- REVEAL 2.0 risk score
- European Society of Cardiology/European Respiratory Society (ESC/ERS) risk stratification
- French Pulmonary Hypertension Registry risk equation
- COMPERA risk assessment
Medical Tests
Hemodynamic Assessment:
- Right Heart Catheterization (Gold Standard):
- Confirms diagnosis with direct pressure measurements
- Defines hemodynamic profile (precapillary vs. postcapillary PH)
- Assesses vasoreactivity for treatment decisions
- Key measurements:
- Mean pulmonary arterial pressure (mPAP)
- Pulmonary capillary wedge pressure (PCWP)
- Pulmonary vascular resistance (PVR)
- Cardiac output/cardiac index
- Mixed venous oxygen saturation
- Vasoreactivity in response to nitric oxide, epoprostenol, or adenosine
Imaging Studies:
Echocardiography:
- First-line non-invasive screening tool
- Estimates pulmonary artery systolic pressure
- Assesses right ventricular size and function
- Evaluates for left heart disease
- Detects congenital heart defects
- Tricuspid regurgitation velocity >2.8 m/s suggests PH
Chest X-ray:
- Enlarged central pulmonary arteries
- Right heart enlargement
- Pruning of peripheral vessels
- May show underlying lung disease
- Normal in early disease stages
Ventilation/Perfusion (V/Q) Scan:
- Essential for CTEPH screening
- Multiple segmental perfusion defects suggest CTEPH
- More sensitive than CT for CTEPH detection
- Normal or subsegmental defects argue against CTEPH
CT Pulmonary Angiography:
- Visualizes thromboembolic disease
- Evaluates lung parenchyma
- Assesses cardiac size and structure
- Measures pulmonary artery diameter
- CT findings suggestive of PH:
- Main pulmonary artery diameter >29 mm
- Ratio of pulmonary artery to aorta diameter >1
- Right ventricular enlargement
- Mosaic attenuation pattern
- Peripheral vessel pruning
High-Resolution CT:
- Evaluates for interstitial lung disease
- Screens for pulmonary veno-occlusive disease
- Identifies emphysema and other lung pathologies
Cardiac MRI:
- Gold standard for right ventricular assessment
- Quantifies right ventricular volumes and function
- Measures stroke volume and cardiac output
- Evaluates for congenital anomalies
- Assesses myocardial fibrosis with late gadolinium enhancement
- Prognostic value through multiple parameters
Pulmonary Angiography:
- Traditional gold standard for CTEPH diagnosis
- Now mainly used during pulmonary endarterectomy planning
- Direct visualization of pulmonary vasculature
- Allows for intervention in selected cases
Laboratory Tests:
Brain Natriuretic Peptide (BNP)/NT-proBNP:
- Biomarkers of cardiac strain
- Elevated in right ventricular pressure overload
- Correlates with disease severity and prognosis
- Useful for monitoring disease progression
Complete Blood Count:
- Screens for polycythemia (chronic hypoxemia)
- Identifies anemia (associated with worse outcomes)
- Assesses for thrombocytopenia
Comprehensive Metabolic Panel:
- Evaluates liver and kidney function
- Electrolyte abnormalities in right heart failure
- Albumin levels (nutritional status)
Thyroid Function Tests:
- Both hypo- and hyperthyroidism can exacerbate PH
- Required before starting specific therapies
HIV Testing:
- HIV-associated PAH is an important subtype
- Part of standard screening
Autoimmune Serologies:
- Antinuclear antibodies (ANA)
- Anti-centromere antibodies
- Anti-SCL-70 antibodies
- Anti-U1-RNP antibodies
- Important for connective tissue disease-associated PAH
Liver Function Tests with Portal Doppler:
- Screening for portopulmonary hypertension
- Assessment of right heart failure impact on liver
Genetic Testing:
- BMPR2, ACVRL1, ENG, CAV1, KCNK3, TBX4
- Recommended for IPAH, heritable PAH, drug-induced PAH
- Cascade family screening when mutations identified
Functional Assessment:
6-Minute Walk Test (6MWT):
- Measures exercise capacity
- Strong prognostic indicator
- Monitors response to therapy
- <300m associated with worse outcomes
Cardiopulmonary Exercise Testing (CPET):
- Gold standard for exercise capacity assessment
- Measures peak oxygen consumption (VO₂)
- Evaluates ventilatory efficiency (VE/VCO₂ slope)
- Differentiates between cardiac and pulmonary limitations
Pulmonary Function Tests:
- Spirometry
- Lung volumes
- Diffusion capacity (often reduced in PAH)
- Arterial blood gases
Sleep Studies:
- Identifies sleep-disordered breathing
- Screens for nocturnal hypoxemia
- Assesses for overlap with pulmonary hypertension
Lung Biopsy:
- Rarely indicated due to high risk
- May be considered in unclear diagnoses
- Can identify pulmonary veno-occlusive disease or pulmonary capillary hemangiomatosis
Early Detection Methods and Effectiveness
Screening Approaches:
Echocardiographic Screening in High-Risk Populations:
- Systemic sclerosis patients: Annual echocardiography recommended
- First-degree relatives of heritable PAH patients
- HIV-infected individuals with unexplained dyspnea
- Congenital heart disease patients with shunts
- Portal hypertension patients being evaluated for liver transplantation
Genetic Screening:
- Cascade genetic testing in families with known mutations
- Genetic counseling for asymptomatic carriers
- 20-40% lifetime risk of developing PAH with BMPR2 mutations
Biomarker Screening:
- NT-proBNP/BNP as part of systemic sclerosis screening
- Emerging role for other biomarkers (growth differentiation factor-15, cardiac troponin)
Effectiveness of Early Detection:
Systemic Sclerosis Screening Programs:
- Earlier detection associated with better functional class at diagnosis
- DETECT algorithm: Two-step algorithm with 96% sensitivity
- Mortality benefit of early detection demonstrated in observational studies
- Reduction in patients diagnosed in WHO Functional Class III-IV
Congenital Heart Disease:
- Regular echocardiographic monitoring detects increased pulmonary pressures before symptom onset
- Early shunt closure prevents Eisenmenger syndrome development
- Clear guidelines for timing of intervention
Limitations of Current Screening:
- Echocardiography has false positives and false negatives
- Right heart catheterization not feasible as initial screening tool
- Most patients still diagnosed at advanced stages
- Cost-effectiveness not established for general population screening
- Asymptomatic early disease often missed
Screening Strategy Effectiveness:
- Targeted screening: Effective in high-risk groups
- Symptom-based screening: Limited by non-specific early symptoms
- Biomarker-based screening: Promising but requires validation
- Combination approaches: Highest yield (e.g., DETECT algorithm)
Early detection of pulmonary hypertension remains challenging due to the non-specific nature of early symptoms and the invasive nature of definitive testing. However, advances in non-invasive assessment and improved risk stratification have enhanced the ability to detect PH earlier in high-risk populations, potentially improving outcomes through earlier intervention.
8. Treatment Options
Standard Treatment Protocols
General Approach: Treatment strategy depends on the WHO group, severity, and individual patient factors. A comprehensive approach includes:
- Treating the underlying cause when possible
- Using supportive therapies for all types
- Implementing specific pulmonary vasodilator therapies for Group 1 PAH and selected patients in other groups
- Regular risk assessment to guide therapy escalation
Treatment Goals:
- Improve symptoms and quality of life
- Enhance exercise capacity
- Normalize right ventricular function
- Improve hemodynamics
- Reduce hospitalizations
- Improve survival
Risk-Based Treatment Approach: Current guidelines recommend a risk-based approach with goal-oriented therapy:
- Low-risk patients: 5-year survival >90%
- Intermediate-risk patients: 5-year survival 50-85%
- High-risk patients: 5-year survival <50%
Treatment is adjusted to maintain or achieve low-risk status.
Medications
General Supportive Therapies (All PH Groups):
Diuretics:
- Loop diuretics (furosemide, torsemide)
- Aldosterone antagonists (spironolactone)
- Manage fluid overload and right heart failure symptoms
Oxygen Supplementation:
- Maintains oxygen saturation >90%
- Reduces hypoxic pulmonary vasoconstriction
- Particularly important in Group 3 PH
Anticoagulants:
- Recommended for CTEPH (Group 4)
- Considered in selected Group 1 PAH patients
- Warfarin or direct oral anticoagulants
- Risk-benefit must be carefully assessed
Iron Supplementation:
- Addresses common iron deficiency
- May improve exercise capacity and quality of life
Specific PAH Therapies (Primarily for Group 1):
Calcium Channel Blockers:
- Limited to the ~5-10% of IPAH patients with positive acute vasoreactivity testing
- High-dose nifedipine, diltiazem, or amlodipine
- Continued only if clinical and hemodynamic improvement demonstrated
Endothelin Receptor Antagonists (ERAs):
- Block endothelin-1, a potent vasoconstrictor
- Ambrisentan: Selective ETA receptor antagonist
- Bosentan: Dual ETA/ETB receptor antagonist
- Macitentan: Tissue-targeting ERA with improved liver safety profile
- Side effects include hepatotoxicity, peripheral edema, anemia
Phosphodiesterase-5 Inhibitors (PDE-5i):
- Increase cyclic GMP, promoting vasodilation
- Sildenafil: Three times daily dosing
- Tadalafil: Once-daily dosing
- Side effects include headache, flushing, nasal congestion
Soluble Guanylate Cyclase Stimulators:
- Directly stimulate guanylate cyclase independent of nitric oxide
- Riociguat: Approved for PAH and inoperable CTEPH
- Side effects include hypotension, dizziness, gastrointestinal symptoms
Prostacyclin Pathway Medications:
- Replace deficient endogenous prostacyclin
- Potent vasodilators with antiproliferative effects
a. Prostacyclin Analogues:
- Epoprostenol: Continuous IV infusion, short half-life
- Treprostinil: Available as IV, subcutaneous, inhaled, and oral formulations
- Iloprost: Inhaled formulation, requires 6-9 doses daily
b. Prostacyclin Receptor Agonist:
- Selexipag: Oral selective IP receptor agonist
- Titrated to maximum tolerated dose
c. Side effects include headache, jaw pain, diarrhea, flushing, and delivery system complications for parenteral forms
Combination Therapy Approaches:
- Initial Combination Therapy: Starting with two medications for intermediate/high-risk patients
- Sequential Combination Therapy: Adding medications for inadequate response
- Triple Combination Therapy: For severe disease or inadequate response to dual therapy
- Most common combinations:
- ERA + PDE-5i
- Prostacyclin + ERA + PDE-5i
- ERA + sGC stimulator
Group-Specific Medical Approaches:
Group 2 (Left Heart Disease):
- Optimize treatment of underlying heart failure/valve disease
- Specific PAH therapies generally not recommended
- Careful diuresis and blood pressure management
Group 3 (Lung Disease):
- Optimize treatment of underlying lung disease
- Oxygen therapy
- Pulmonary rehabilitation
- Selected patients may benefit from specific PAH therapies
Group 4 (CTEPH):
- Lifelong anticoagulation
- Riociguat for inoperable disease or residual PH after surgery
- Other PAH medications may be considered off-label
- Balloon pulmonary angioplasty for selected patients
Group 5 (Unclear Mechanisms):
- Treat underlying disorder
- PAH therapies based on individual assessment
Surgical and Interventional Options
Surgical Approaches:
Pulmonary Endarterectomy (PEA):
- Curative procedure for CTEPH
- Surgical removal of organized thromboembolic material
- Should be considered in all eligible CTEPH patients
- In-hospital mortality <5% at expert centers
- Significant improvements in hemodynamics and survival
Lung Transplantation:
- Option for advanced disease unresponsive to medical therapy
- Bilateral transplantation preferred
- 5-year survival 45-60%
- Limited by organ availability
- Candidate selection critical for success
Heart-Lung Transplantation:
- Reserved for Eisenmenger syndrome with complex cardiac defects
- Significant end-organ dysfunction
- Limited availability and higher complexity
Atrial Septostomy:
- Palliative procedure creating right-to-left shunt
- Decompresses right heart chambers
- Improves left ventricular filling and cardiac output
- Bridge to transplantation
- High procedural risk
Potts Shunt:
- Direct connection between left pulmonary artery and descending aorta
- Creates partial right-to-left shunt
- Preserves upper body oxygenation
- Predominantly used in pediatric patients
Interventional Approaches:
Balloon Pulmonary Angioplasty (BPA):
- Emerging option for inoperable CTEPH
- Series of staged procedures
- Mechanical disruption of organized thrombi
- Significant hemodynamic improvements
- Specialized centers report outcomes approaching surgical results
Percutaneous Balloon Atrial Septostomy:
- Less invasive alternative to surgical septostomy
- Staged approach possible
- Careful patient selection essential
Pulmonary Artery Denervation:
- Investigational procedure
- Targets sympathetic nervous system
- Initial studies show hemodynamic improvements
- Long-term benefits unclear
Emerging Treatments and Clinical Trials
Novel Pharmacological Approaches:
Tyrosine Kinase Inhibitors:
- Imatinib showed hemodynamic improvements but safety concerns
- Second-generation TKIs with improved safety profiles under investigation
- Target proliferative aspect of vascular remodeling
Rho Kinase Inhibitors:
- Fasudil and newer compounds
- Address both vasoconstriction and vascular remodeling
- Phase 2 studies ongoing
Inflammation-Targeted Therapies:
- Rituximab for connective tissue disease-associated PAH
- Tocilizumab (IL-6 receptor antagonist)
- TNF-alpha inhibitors
Metabolic Modulators:
- Dichloroacetate: Targets mitochondrial dysfunction
- Ranolazine: Improves right ventricular energetics
- Trimetazidine: Metabolic efficiency enhancer
BMPR2-Enhancing Strategies:
- FK506 (tacrolimus): Increases BMPR2 signaling
- Elafin: Protects against elastase-mediated BMPR2 degradation
Cellular and Gene Therapies:
Mesenchymal Stem Cell Therapy:
- Early-phase clinical trials show promise
- Anti-inflammatory and regenerative effects
- Investigate multiple delivery approaches
Endothelial Progenitor Cells:
- Autologous or allogeneic
- May promote vascular repair
- Phase 1/2 trials underway
Gene Therapy:
- BMPR2 gene delivery
- Micro-RNA modulation
- Adeno-associated virus vectors
- Preclinical and early clinical investigation
Device-Based Approaches:
Right Ventricular Assist Devices:
- Modified for pulmonary circulation support
- Bridge to transplant or recovery
- Technical challenges being addressed
Pumpless Lung Assist:
- Novalung and similar devices
- Reduce right ventricular afterload
- Oxygenate blood
- Primarily as bridge to transplant
Selected Notable Clinical Trials:
- Phase 3 trials of novel combination approaches
- Head-to-head comparisons of treatment strategies
- Trials targeting specific PH subtypes (e.g., scleroderma-associated PAH)
- Studies of early intervention in asymptomatic BMPR2 carriers
- Trials of non-invasive wearable hemodynamic monitoring
- Studies addressing optimal timing for lung transplantation
- Trials focusing on right ventricular function preservation
Treatment of pulmonary hypertension has evolved dramatically over the past two decades, transforming prognosis for many patients. However, current therapies primarily slow progression rather than cure the disease, and the development of truly transformative approaches remains an area of intense research.
9. Prevention & Precautionary Measures
Primary Prevention Strategies
Prevention strategies vary by PH classification group and underlying risk factors:
General Population Measures:
- Avoid known risk factors (appetite suppressants, methamphetamines)
- Maintain healthy weight
- Regular physical activity
- Smoking cessation
- Prompt treatment of sleep apnea
Group-Specific Prevention:
Group 1 (PAH):
- Avoid anorexigen drugs
- Screening in high-risk individuals (systemic sclerosis, HIV)
- Genetic counseling for families with heritable PAH
- Careful monitoring during pregnancy in at-risk women
- Caution with certain chemotherapeutic agents (dasatinib)
Group 2 (Left Heart Disease):
- Optimal management of heart failure and valvular disease
- Blood pressure control
- Early valve intervention when indicated
- Cardiac rehabilitation
- Dietary sodium restriction
Group 3 (Lung Disease/Hypoxia):
- Smoking cessation programs
- COPD/asthma management
- Supplemental oxygen when indicated
- Vaccination against respiratory infections
- Pulmonary rehabilitation
- Prevention of occupational lung disease
Group 4 (CTEPH):
- Appropriate anticoagulation for acute pulmonary embolism
- Adequate duration of therapy for provoked/unprovoked PE
- Screening for thrombophilia in selected cases
- Follow-up after significant PE to detect persistent symptoms
Group 5 (Multifactorial):
- Targeted to specific underlying cause
- Regular monitoring in sarcoidosis, hematological disorders
Prevention of Known Risk Factors:
- Regulation of appetite suppressants
- Workplace safety measures for chemical exposure
- Avoiding high-altitude exposure in susceptible individuals
- Prompt treatment of autoimmune conditions
- Screening before initiating potentially harmful drugs
Lifestyle Modifications and Environmental Precautions
For Patients Already Diagnosed:
Physical Activity Recommendations:
- Regular, low-level aerobic activity encouraged
- Avoid activities causing excessive breathlessness
- Supervised pulmonary rehabilitation programs
- Avoid heavy isometric exercises
- Avoid high-altitude exercise
- Oxygen supplementation during activity if needed
Nutritional Considerations:
- Sodium restriction (2000-2400 mg/day) for right heart failure
- Adequate protein intake to maintain muscle mass
- Iron-rich foods or supplementation
- Vitamin D and calcium for bone health
- Avoid excessive fluid intake with right heart failure
- Small, frequent meals if early satiety present
Travel Considerations:
- Commercial air travel generally safe with supplemental oxygen if needed
- Avoid high-altitude destinations (>1500-2000m) without consultation
- Medical oxygen arrangements for travel
- Thromboprophylaxis for long journeys
- Carry medication documentation and emergency contact information
- Travel insurance covering pre-existing conditions
Environmental Precautions:
- Indoor air quality management
- Avoiding extreme temperatures
- Air conditioning during hot weather
- Caution with air pollution exposure
- Avoid wood smoke and other respiratory irritants
- Proper ventilation in workplace
Stress Management:
- Psychological support
- Relaxation techniques
- Cognitive behavioral therapy
- Support groups
- Adequate sleep hygiene
Preventive Monitoring and Screening
Monitoring in High-Risk Populations:
Systemic Sclerosis Patients:
- Annual echocardiography
- DETECT algorithm application (combines clinical, laboratory, and echo parameters)
- Right heart catheterization if screening suggests PH
- Earlier intervention improves outcomes
BMPR2 Mutation Carriers:
- Regular echocardiographic surveillance
- Low threshold for further evaluation with symptoms
- Exercise echocardiography to detect early abnormalities
- Genetic counseling regarding transmission risks
First-Degree Relatives of PAH Patients:
- Consider genetic testing if mutation identified in family
- Clinical screening with echocardiography
- Education about early symptoms
Congenital Heart Disease Patients:
- Regular monitoring based on defect type
- Early intervention for shunt lesions
- Particularly important in unrepaired or partially repaired defects
HIV Patients:
- Echocardiographic screening with unexplained dyspnea
- Annual cardiopulmonary evaluation recommended by some experts
Post-Pulmonary Embolism Patients:
- Follow-up evaluation for persistent symptoms
- V/Q scanning or CTPA if symptoms persist
- Exercise capacity assessment
Preventive Vaccination:
- Annual influenza vaccination
- Pneumococcal vaccination
- COVID-19 vaccination
- Respiratory syncytial virus prevention in selected patients
- General age-appropriate vaccinations
Prophylactic Strategies:
- Consideration of prophylactic anticoagulation in high-risk settings
- Early mobilization after surgery
- Compression stockings for venous stasis
- Hydration maintenance
- Thromboprophylaxis during pregnancy in high-risk women
While primary prevention of pulmonary hypertension remains challenging, targeted screening of high-risk populations and modification of risk factors can potentially identify disease at earlier stages when treatment is more effective. The multifactorial nature of most forms of PH necessitates a comprehensive approach to prevention spanning multiple health domains.
10. Global & Regional Statistics
Global Incidence and Prevalence
Overall Pulmonary Hypertension:
- Global prevalence estimates: 1% of global population (~78 million people)
- Significant underdiagnosis likely in resource-limited regions
- Increasing prevalence with aging populations
- Detection rates increasing with improved diagnostic tools
Group 1 (PAH) Statistics:
- Global prevalence: 15-60 cases per million population
- Annual incidence: 2-7.6 cases per million per year
- Registration data suggests rising incidence
- Idiopathic PAH: 5-10 cases per million
- Heritable PAH: 1-3 cases per million
- Drug/toxin-induced: Variable based on regional exposures
- Associated PAH:
- CTD-associated: 3-15 cases per million
- HIV-associated: 0.5% of HIV patients
- Portopulmonary: 2-6% of patients with portal hypertension
- CHD-associated: 5-10% of adults with congenital heart disease
Group 2 (Left Heart Disease):
- Most common form globally (65-80% of all PH cases)
- Prevalence in heart failure with reduced ejection fraction (HFrEF): 40-75%
- Prevalence in heart failure with preserved ejection fraction (HFpEF): 36-83%
- Prevalence in valvular heart disease: 26-55% of mitral stenosis cases
Group 3 (Lung Disease/Hypoxia):
- Second most common form globally
- Prevalence in COPD: 30-70% (severe PH in 3-5%)
- Prevalence in interstitial lung disease: 30-60%
- Prevalence in sleep-disordered breathing: 17-53%
- High-altitude PH: 5-18% of populations living above 3000m
Group 4 (CTEPH):
- Global prevalence: 3-30 per million
- Cumulative incidence after acute PE: 0.1-9.1% within 2 years
- Likely underdiagnosed in many regions
- Higher detection rates in countries with CTEPH treatment centers
Group 5 (Multifactorial Mechanisms):
- Limited global data
- Prevalence in sarcoidosis: 5-20% of all cases
- Myeloproliferative disorder-associated: 10-48% in primary myelofibrosis
Regional Variations in Prevalence
North America:
- PAH registry data estimates 12-50 cases per million
- Highest proportion of idiopathic and heritable PAH
- Significant racial disparities in access to diagnosis and treatment
- Higher rates of obesity and sleep apnea contributing to Group 2 and 3 PH
Europe:
- PAH prevalence: 15-60 per million
- National registries document variations between countries
- Highest rates of CTD-associated PAH in Sweden and UK
- Higher rates of CTEPH diagnosis due to specialized centers
- French PAH registry: 15 cases per million
- UK National Audit: 52 cases per million
Asia:
- Variable detection rates based on healthcare infrastructure
- Japan: 17 cases of PAH per million
- China: Lower reported prevalence (possibly due to underdiagnosis)
- Higher rates of CHD-associated PAH in regions with delayed CHD repair
- Significant CTEPH burden in Japan with specialized treatment programs
Latin America:
- Limited systematic data
- Higher rates of schistosomiasis-associated PAH in Brazil and certain regions
- Higher rates of high-altitude PH in Andean regions
- Challenges in access to advanced therapies
Africa:
- Significant underdiagnosis and limited data
- Higher rates of HIV-associated PAH
- Schistosomiasis-associated PAH in endemic regions
- Rheumatic heart disease contributing to Group 2 PH
- Limited access to diagnostic tools and therapies
Australia/Oceania:
- PAH prevalence similar to Europe/North America
- Better characterization due to centralized healthcare systems
- National registries tracking outcomes
- Lower rates of certain etiologies (schistosomiasis, high-altitude)
Mortality and Survival Rates
Historical vs. Current Survival:
- Historical (pre-specific therapy) median survival for IPAH: 2.8 years
- Modern treated PAH survival rates:
- 1-year survival: 85-93%
- 3-year survival: 68-80%
- 5-year survival: 55-75%
- 7-year survival: 45-60%
Survival by PH Classification:
- Group 1 PAH: 5-year survival 55-75% with modern therapy
- Group 2 PH: Heavily influenced by underlying cardiac disease (5-year survival 30-70%)
- Group 3 PH: 5-year survival 30-50% (worse with severe PH)
- Group 4 CTEPH:
- After successful PEA: 5-year survival 80-90%
- Inoperable treated with medical therapy: 5-year survival 55-70%
- Untreated: 3-year survival <40%
- Group 5: Highly variable depending on underlying cause
Survival by PAH Subtype:
- Idiopathic/Heritable PAH: 5-year survival 65-70%
- CTD-associated PAH: 5-year survival 45-60%
- HIV-associated PAH: 5-year survival 60-65%
- Portopulmonary: 5-year survival 40-45%
- CHD-associated (Eisenmenger): 5-year survival 75-90%
Regional Differences in Survival:
- Highest survival rates in North America, Western Europe, Japan, Australia
- Lower survival in Eastern Europe, parts of Asia, Africa, South America
- Direct correlation with access to specialized care centers and specific therapies
- 5-year survival variation: 45-85% depending on region and healthcare access
Mortality Causes in PAH:
- Progressive right heart failure: 30-50%
- Sudden death: 25-30%
- Respiratory failure/pneumonia: 10-15%
- Comorbidity-related deaths: 10-20%
- Treatment complications: 5-10%
Global Economic Burden and Trends
Healthcare Costs:
- Average annual direct healthcare costs per PAH patient:
- United States: $75,000-$116,000
- Europe: €40,000-€80,000
- Asia: Variable based on country and access to therapies
- Main cost drivers:
- PAH-specific medications (55-75% of costs)
- Hospitalizations (15-25%)
- Diagnostic procedures (5-10%)
- Outpatient care (5-15%)
Disability and Indirect Costs:
- Lost productivity: €4,000-€10,000 annually per patient in Europe
- Caregiver burden: €8,000-€15,000 annual estimated value
- Disability rates: 35-75% of PAH patients unable to work
- Total economic burden (direct + indirect) in US: Estimated >$4.5 billion annually
Access to Care Disparities:
- Only 30% of countries globally have specialized PH treatment centers
- <40% of countries have access to all classes of PAH-specific therapies
- 7% of low-income countries have access to prostacyclin therapies
- 60% of patients globally estimated to receive suboptimal therapy
Current Trends:
- Increasing diagnosis rates globally
- Earlier detection through improved screening protocols
- Aging of the PH population with improved survival
- Growing burden of Group 2 and 3 PH with population aging
- Decreasing mortality with combination therapy approaches
- Geographic expansion of specialized care centers
- Increasing cost pressure on healthcare systems from expensive therapies
- Emerging generic options potentially improving access
Pulmonary hypertension represents a substantial global health burden with marked disparities in diagnosis, treatment access, and outcomes. While advances in therapeutic options have dramatically improved survival in well-resourced healthcare settings, significant challenges remain in ensuring equitable access to diagnosis and treatment worldwide.
11. Recent Research & Future Prospects
Latest Advancements in Treatment and Research
Therapeutic Innovations:
Novel Drug Delivery Systems:
- Implantable pumps for continuous prostacyclin delivery
- Extended-release oral treprostinil formulations
- Inhaled dry-powder treprostinil reducing administration burden
- Subcutaneous pump systems with longer reservoir life
Combination Therapy Optimization:
- AMBITION trial established upfront dual combination therapy as standard of care
- Triple therapy approaches showing promise in advanced disease
- Personalized combination strategies based on patient phenotypes
- Sequential add-on vs. upfront combination approaches being refined
Precision Medicine Approaches:
- Genetic testing to guide therapy selection
- Biomarker-based treatment decisions
- Responder phenotype identification
- Individualized risk assessment tools
- Machine learning algorithms for treatment response prediction
Interventional Advances:
- Refinement of balloon pulmonary angioplasty techniques
- Transcatheter potts shunt creation
- Atrial flow regulator devices for controlled shunting
- Pulmonary artery denervation procedures
- 3D mapping of pulmonary circulation for intervention planning
Diagnostic Advancements:
Imaging Innovations:
- 4D flow MRI characterizing pulmonary circulation
- PET imaging for pulmonary vascular inflammation
- Strain echocardiography for early right ventricular dysfunction detection
- CT-derived fractional flow reserve measurements
- Dual-energy CT improving CTEPH detection
Biomarker Development:
- Multi-marker panels (combining natriuretic peptides, troponin, other markers)
- MicroRNA signatures specific to PAH
- Metabolomic profiling
- Volatile organic compounds in exhaled breath
- Circulating endothelial cells and microparticles
Risk Stratification Tools:
- REVEAL 2.0 risk calculator refinement
- ESC/ERS simplified risk assessment
- Integration of imaging and biomarker data
- Machine learning-based prediction models
- Dynamic risk assessment protocols
Basic Science Discoveries:
Molecular Pathway Advances:
- Deeper understanding of BMPR2 signaling networks
- Recognition of metabolic abnormalities in pulmonary vasculature
- DNA damage response pathways in PAH
- Epigenetic modifications contributing to disease
- Role of microRNAs in regulating pulmonary vascular function
Cellular Interactions:
- Endothelial-smooth muscle cell crosstalk
- Immune cell contributions to vascular remodeling
- Role of pericytes in pulmonary vascular homeostasis
- Fibroblast activation in PH development
- Platelet abnormalities and vascular dysfunction
Genetic/Genomic Insights:
- Expansion of PAH-associated gene panels (now >20 genes)
- Whole genome sequencing identifying new genetic contributors
- Insights into genetic penetrance modulators
- Polygenic risk scores in development
- Genetic modifiers explaining clinical heterogeneity
Ongoing Studies and Future Directions
Major Ongoing Clinical Trials:
Novel Therapeutic Targets:
- Sotatercept (activin receptor fusion protein) in phase 3 trials
- LOXL2 inhibitors targeting vascular fibrosis
- Apelin receptor agonists
- Serotonin pathway modulators
- CXC chemokine receptor antagonists
Treatment Strategy Trials:
- Early combination therapy vs. sequential therapy
- Risk-based treatment algorithms
- Withdrawal protocols for stabilized patients
- Comparative effectiveness studies of different combinations
- Pulsed upfront intensive therapy concepts
Right Ventricular Focus:
- Beta-blocker use in stabilized PAH patients
- Ranolazine for right ventricular diastolic dysfunction
- Cardiac rehabilitation specific to RV dysfunction
- Mechanical circulatory support approaches
- Mitochondrial metabolic modulators
Subgroup-Specific Approaches:
- Scleroderma-PAH targeted immunomodulation
- HIV-associated PAH specialized protocols
- PVOD/PCH targeted therapies
- Pediatric PAH optimized approaches
- Elderly-onset PAH management
Future Research Directions:
Disease Modification:
- Targeting vascular remodeling and regeneration
- Anti-inflammatory approaches
- Reversal of established pathology
- Prevention strategies in high-risk groups
- Epigenetic modifying agents
Right Ventricular Focus:
- RV-directed therapies beyond afterload reduction
- Enhancing RV-pulmonary artery coupling
- Contractility enhancement approaches
- RV fibrosis prevention
- RV metabolic modulation
Technology Integration:
- Wearable hemodynamic monitoring
- Artificial intelligence for image analysis
- Remote patient monitoring systems
- Decision support systems for physicians
- Digital therapeutics for symptom management
System-Based Improvements:
- Care delivery model optimization
- Telehealth integration for PH management
- Shared decision-making tools
- Patient-reported outcome implementation
- Global access expansion strategies
Potential Cures and Innovative Approaches
Regenerative Medicine Approaches:
Stem Cell Therapies:
- Endothelial progenitor cells for vascular repair
- Mesenchymal stem cells for anti-inflammatory effects
- Induced pluripotent stem cell-derived endothelial cells
- Engineered cell therapy platforms
- Exosome and microvesicle therapeutic approaches
Gene Therapy:
- BMPR2 gene delivery systems
- Gene editing for heritable PAH mutations
- MicroRNA-based therapeutics
- Nanoparticle delivery systems
- Epigenetic editing approaches
Innovative Treatment Paradigms:
Pulmonary Vascular Remodeling Reversal:
- Combined anti-proliferative and pro-apoptotic approaches
- Elastase inhibition to restore vascular compliance
- Targeted drug delivery to pulmonary circulation
- Decellularized lung matrix approaches
- Bioengineered vascular grafts
Metabolism-Targeting Approaches:
- Mitochondrial function enhancers
- Glycolysis inhibitors reversing Warburg effect
- Glutaminolysis modulation
- Fatty acid oxidation enhancement
- Dietary approaches to vascular metabolism
Immune System Modulation:
- Regulatory T-cell amplification
- B-cell targeted therapies in autoimmune-associated PAH
- Innate immunity modulation
- Mast cell stabilization approaches
- Targeted anti-inflammatory cytokine therapies
Next-Generation Technologies:
Advanced Interventional Approaches:
- Percutaneous pulmonary vascular stenting for CTEPH
- Image-guided targeted drug delivery
- Biodegradable pulmonary arterial support devices
- Radio-frequency ablation of sympathetic nerves
- Pulmonary artery pressure modulation devices
Personalized and Precision Medicine:
- Comprehensive genetic and phenotypic profiling
- Real-time therapy adjustment based on hemodynamic monitoring
- Patient-specific cell models for drug testing
- Digital twins for therapy simulation
- Multi-omics approach to treatment selection
Artificial Intelligence Applications:
- Early detection algorithms from electronic health records
- Imaging analysis for subtle RV dysfunction
- Predictive modeling for treatment response
- Drug repurposing through computational approaches
- Natural language processing of clinical notes for risk factors
While a definitive cure for pulmonary hypertension remains elusive, the pace of discovery has accelerated dramatically. The shift from single-target approaches to comprehensive strategies addressing multiple disease pathways offers hope for transformative therapy. The combination of genetic insights, technological advances, and deepening understanding of disease mechanisms may ultimately lead to disease modification or prevention strategies, particularly for genetically predisposed individuals.
12. Interesting Facts & Lesser-Known Insights
Uncommon Knowledge About Pulmonary Hypertension
Historical Perspectives:
- The first clinical description of pulmonary hypertension dates back to 1891, but the ability to actually measure pulmonary pressures wasn’t possible until cardiac catheterization was developed in the 1940s
- The “aminorex epidemic” of PAH in the 1960s in Austria, Germany, and Switzerland was the first recognition that drugs could cause PAH, when 1 in 500 users of this appetite suppressant developed the condition
- The term “primary pulmonary hypertension” was used until 1998, when the more precise classification system was developed
- The first truly effective treatment (epoprostenol) wasn’t approved until 1995, less than 30 years ago
Biological Curiosities:
- The pulmonary circulation normally operates as a high-flow, low-pressure, low-resistance system, handling the same cardiac output as the systemic circulation but at 1/5 the pressure
- The pulmonary vasculature contains about 1/10 the amount of smooth muscle as systemic vessels, which is why it’s so sensitive to vasoconstrictive stimuli
- Pulmonary vessels constrict in response to hypoxia (low oxygen), the opposite response to systemic vessels – this improves ventilation-perfusion matching but contributes to altitude-related PH
- The right ventricle has approximately 1/3 the muscle mass of the left ventricle and adapts poorly to pressure overload
- PAH tissues show cancer-like metabolic changes (Warburg effect) with shift to aerobic glycolysis
Clinical Pearls:
- The time from symptom onset to diagnosis averages 2-3 years due to the non-specific nature of early symptoms
- 40% of PAH patients may have evidence of right ventricular ischemia despite normal coronary arteries, due to increased oxygen demand and reduced perfusion
- Iron deficiency affects up to 63% of PAH patients and contributes to worse outcomes, even without anemia
- Pericardial effusion, a collection of fluid around the heart, is one of the strongest predictors of poor survival in PAH
- Patients with PAH can develop compression of the left main coronary artery by an enlarged pulmonary artery, causing angina-like symptoms
Physiological Insights:
- The right ventricle can increase its output five-fold during exercise in healthy individuals
- Pulmonary vascular resistance normally decreases during exercise, unlike in PH patients where it may increase
- Up to 30% of the general population has a patent foramen ovale that may open during high pulmonary pressures, causing right-to-left shunting
- Vocal cord paralysis can occur in severe PAH due to compression of the left recurrent laryngeal nerve by the enlarged pulmonary artery (Ortner’s syndrome)
- The pulmonary circulation contains approximately 10% of the total blood volume, serving as both a conduit and a reservoir
Myths and Misconceptions vs. Medical Facts
Myth: Pulmonary hypertension is just high blood pressure affecting the lungs. Fact: While both involve elevated pressure, pulmonary hypertension is a complex vascular disease with significant remodeling of blood vessels, not simply elevated pressure like systemic hypertension.
Myth: All patients with pulmonary hypertension should avoid exercise. Fact: Supervised, appropriate exercise is actually beneficial for most PH patients and is recommended in stable patients. Extreme exertion should be avoided, but regular low-level activity improves quality of life and may stabilize or improve right heart function.
Myth: Pulmonary hypertension primarily affects older adults. Fact: While some forms are more common in older individuals, PAH has a peak incidence in the third to fourth decade of life, affecting many young adults in their most productive years.
Myth: Supplemental oxygen isn’t necessary unless a patient feels short of breath. Fact: Chronic hypoxemia can worsen pulmonary hypertension even when not causing noticeable symptoms. Oxygen therapy should be guided by measured oxygen levels, not just symptoms.
Myth: Calcium channel blockers are effective treatments for most PH patients. Fact: Only about 5-10% of PAH patients respond to calcium channel blockers, specifically those who show vasoreactivity during right heart catheterization testing. For non-responders, these medications may be harmful.
Myth: Pulmonary hypertension always causes bluish lips or fingers (cyanosis). Fact: Cyanosis is a late finding primarily seen in advanced disease or in patients with right-to-left shunting. Many patients, even with severe disease, never develop visible cyanosis.
Myth: All treatments for pulmonary hypertension are expensive specialty medications. Fact: While PAH-specific therapies are costly, many supportive treatments (diuretics, oxygen, exercise programs) are relatively inexpensive and provide significant symptom benefit.
Myth: Pregnancy is always fatal in pulmonary hypertension. Fact: While pregnancy carries a high risk in PAH (maternal mortality 16-30%), with modern management at specialized centers, most women can survive pregnancy. However, it remains contraindicated due to significant risks.
Myth: Pulmonary hypertension can be cured with the right medication. Fact: Current treatments can significantly improve symptoms and prognosis, but they control rather than cure the disease for most patients. Only pulmonary endarterectomy for CTEPH offers potential cure.
Impact on Specific Populations and Professions
Pregnancy and Reproductive Health:
- Maternal mortality risk: 16-30% in PAH despite modern management
- Peak risk period: Peripartum and early postpartum period (hemodynamic shifts)
- Contraception essential: Dual methods recommended (hormonal plus barrier)
- Estrogen-containing contraceptives contraindicated in most cases
- Genetic counseling needed for heritable forms: 50% transmission risk of mutations
High-Altitude Populations:
- Chronic mountain sickness (Monge’s disease) affects 5-18% of high-altitude residents
- Pulmonary artery pressure increases linearly with altitude
- Genetic adaptations in Andean, Tibetan and Ethiopian highland populations
- Acetazolamide helps prevent altitude-related pulmonary hypertension
- Permanent residents show vascular remodeling different from acute exposure
Professional Athletes:
- Need for careful evaluation before clearance for competitive sports
- Reduced exercise capacity often first manifestation in athletes
- Static (isometric) exercise particularly problematic in PH
- Altitude training may unmask or worsen underlying PH
- Difficulty distinguishing physiologic RV adaptation from pathology
Occupational Impact:
- High-physical-demand occupations often unsustainable
- Commercial pilots: Disqualified with PAH diagnosis due to risk of incapacitation
- Divers: Pulmonary hypertension contraindicates scuba diving
- Military service restrictions with even mild PH
- Mining, high-altitude work sites may require special consideration
Age-Specific Considerations:
Pediatric PH:
- Different etiology profile: More congenital heart disease, developmental lung disorders
- Growth and development concerns with chronic illness
- Medication dosing challenges
- School attendance and socialization issues
- Transition to adult care critical period
Elderly-Onset PH:
- Multiple comorbidities complicating diagnosis and treatment
- Drug interactions more common
- Reduced physiological reserve
- Diagnostic confusion with heart failure with preserved ejection fraction
- Underrepresentation in clinical trials
Psychosocial Impact:
- Invisible disability aspects: Normal appearance despite severe limitation
- Anxiety and depression in 35-55% of patients
- Social isolation due to activity limitations
- Financial burden of disease management
- Relationship strain and changed family dynamics
- Body image issues with medication devices (pumps, oxygen)
- Difficulty navigating disability systems for “invisible” illness
The complexity of pulmonary hypertension extends beyond its physiological impact, affecting nearly every aspect of patients’ lives. The perspectives and needs of specific patient populations highlight the importance of patient-centered, holistic care approaches that address not only the vascular pathology but also the broader life impact of this challenging condition.
This comprehensive report on pulmonary hypertension reflects current medical understanding as of April 2025. While extensive and evidence-based, it should be used for informational purposes and not as a substitute for personalized medical advice.