
⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
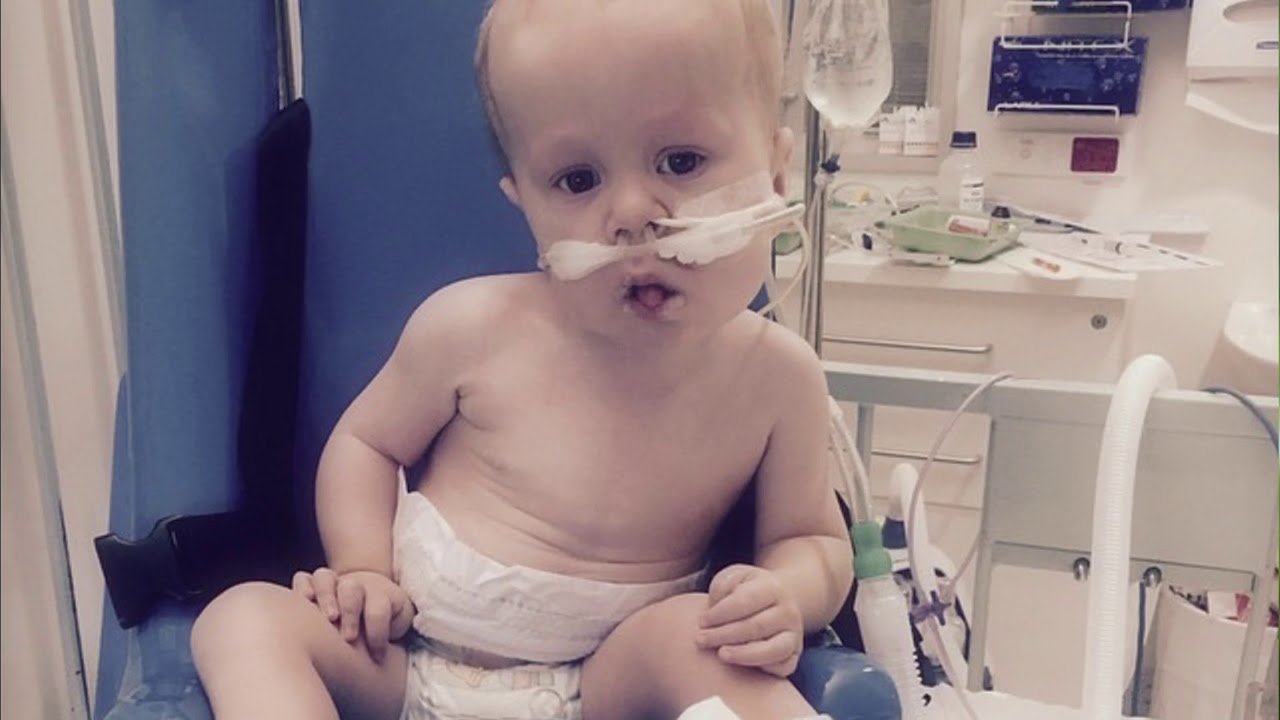
Comprehensive Report on Pompe Disease
1. Overview
Pompe disease, also known as glycogen storage disease type II (GSD-II) or acid maltase deficiency, is a rare, inherited metabolic disorder characterized by the deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is essential for breaking down glycogen (a complex sugar) into glucose within lysosomes, specialized compartments in cells. Without sufficient enzyme activity, glycogen accumulates to toxic levels primarily in muscle cells, causing progressive muscle weakness and damage.
The disease primarily affects cardiac, respiratory, and skeletal muscles throughout the body. The accumulation of glycogen in heart and skeletal muscle cells leads to the enlargement of these organs and progressive muscle weakness. Particularly affected are the heart, liver, nervous system, and muscles used for breathing and movement.
Globally, Pompe disease affects approximately 1 in 40,000 births, though prevalence varies significantly by ethnicity and geographic region. As one of the 50+ lysosomal storage disorders, it represents a significant rare disease with severe health implications when left untreated.
2. History & Discoveries
Pompe disease was first identified in 1932 by Dutch pathologist Dr. Johannes C. Pompe, who described the case of a 7-month-old infant who died from what appeared to be idiopathic hypertrophic cardiomyopathy. During the autopsy, Dr. Pompe discovered massive glycogen accumulation in the infant’s tissues, particularly in the heart muscle. This discovery established the connection between glycogen storage and cardiomegaly in what would later be named after him.
Key historical developments include:
- 1932: First clinical description by Dr. Johannes Pompe
- 1954: Christian de Duve discovered lysosomes, later proving crucial to understanding the disease mechanism
- 1963: H.G. Hers identified the specific enzyme deficiency (acid alpha-glucosidase) responsible for the condition, classifying it as the first recognized lysosomal storage disorder
- 1970s-1980s: Development of diagnostic tests measuring GAA enzyme activity
- 1986: The GAA gene was cloned and mapped to chromosome 17
- 1990s: Initial attempts at enzyme replacement therapy (ERT)
- 2006: FDA approval of alglucosidase alfa (Myozyme) as the first specific treatment for Pompe disease
- 2010: FDA approval of Lumizyme for late-onset Pompe disease
The understanding of Pompe disease evolved from a uniformly fatal condition to a manageable chronic disease through these scientific breakthroughs, particularly the development of enzyme replacement therapy.
3. Symptoms
Pompe disease presents with a spectrum of symptoms varying by age of onset, rate of progression, and degree of organ involvement. The condition is typically classified into two main forms:
Infantile-onset Pompe disease (IOPD):
- Appears within the first months of life
- Profound muscle weakness (hypotonia) or “floppy baby” syndrome
- Enlarged heart (cardiomegaly) with cardiac failure
- Breathing difficulties due to weakened respiratory muscles
- Feeding problems and failure to thrive
- Enlarged tongue (macroglossia)
- Enlarged liver (hepatomegaly)
- Without treatment, often fatal within the first year of life
Late-onset Pompe disease (LOPD):
- Can appear anytime from late childhood to adulthood
- Progressive proximal muscle weakness (affecting hips, thighs, shoulders)
- Gradual respiratory muscle weakness leading to breathing problems
- Sleep-disordered breathing and sleep apnea
- Exercise intolerance and fatigue
- Difficulty rising from chairs, climbing stairs, or lifting objects
- Scoliosis and postural changes
- Usually spares the heart
As the disease progresses, both forms can lead to:
- Worsening respiratory function, potentially requiring ventilatory support
- Loss of mobility and independence
- Swallowing difficulties (dysphagia)
- Voice changes and speech difficulties
- Headaches due to respiratory insufficiency
- Morning headaches and daytime sleepiness from sleep-disordered breathing
While the infantile form progresses rapidly over months, the late-onset form typically progresses more slowly over years or decades.
4. Causes
Pompe disease is caused by mutations in the GAA gene located on chromosome 17q25.2-q25.3. This gene provides instructions for producing the enzyme acid alpha-glucosidase, which is essential for breaking down glycogen in lysosomes.
Genetic Basis:
- Autosomal recessive inheritance pattern, meaning both parents must pass a mutated copy of the gene for a child to be affected
- More than 500 different disease-causing mutations have been identified in the GAA gene
- Different mutations lead to varying levels of residual enzyme activity, correlating with disease severity and age of onset
- Infantile-onset is associated with mutations causing little to no enzyme activity (<1%)
- Late-onset is associated with mutations allowing some residual enzyme activity (1-30%)
Pathophysiology:
- Reduced or absent GAA enzyme activity leads to glycogen accumulation in lysosomes
- This accumulation causes lysosomal swelling and rupture
- Cellular damage occurs, particularly in cardiac, skeletal, and smooth muscle tissues
- Autophagy (cellular recycling process) is disrupted
- Abnormal mitochondrial function further compromises energy production in cells
Unlike some genetic disorders, there are no known environmental triggers or exposures that cause Pompe disease or exacerbate its progression. The disease is solely determined by genetic factors.
5. Risk Factors
As a genetic disorder, the primary risk factors for Pompe disease are related to heredity and family history rather than lifestyle or environmental exposures.
Primary Risk Factors:
- Family history of Pompe disease
- Parents who are both carriers of GAA gene mutations (each having one mutated copy)
- Consanguineous marriage (related parents), which increases the chance of both parents carrying the same rare mutation
Demographic Factors:
- Certain ethnic backgrounds show higher prevalence:
- African Americans (estimated 1:14,000)
- People of Dutch ancestry (approximately 1:40,000)
- Chinese population in Taiwan (approximately 1:50,000)
- Certain isolated populations with founder effects or higher rates of consanguinity
Carrier Frequency:
- Estimated carrier frequency in the general population is about 1:100
- Higher carrier frequencies in specific populations correlating with increased disease prevalence
Since Pompe disease is genetic in nature, there are no known modifiable risk factors such as lifestyle choices, occupational exposures, or pre-existing conditions that increase the risk of developing the disease. However, certain pre-existing conditions may compound the effects of Pompe disease once it manifests.
6. Complications
Pompe disease can lead to numerous complications affecting quality of life and long-term survival:
Respiratory Complications:
- Progressive respiratory insufficiency (leading cause of death)
- Recurrent pneumonia due to weakness in respiratory muscles
- Sleep apnea and sleep-disordered breathing
- Respiratory failure requiring ventilatory support
- Reduced lung capacity and function
Cardiac Complications (primarily in infantile form):
- Hypertrophic cardiomyopathy
- Conduction abnormalities and arrhythmias
- Cardiac failure
- Sudden cardiac death
Musculoskeletal Complications:
- Progressive loss of mobility
- Muscle contractures and joint deformities
- Scoliosis and postural abnormalities
- Osteopenia and increased fracture risk
Gastrointestinal Complications:
- Difficulty swallowing (dysphagia)
- Risk of aspiration pneumonia
- Delayed gastric emptying
- Reduced gut motility
Other Complications:
- Hearing loss
- Arterial aneurysms (particularly basilar artery)
- Chronic pain
- Depression and anxiety related to chronic disease burden
- Reduced quality of life and psychosocial impacts
Mortality Rates:
- Without treatment, infantile-onset Pompe disease is typically fatal before age 2, primarily due to cardiorespiratory failure
- With treatment, 5-year survival rates for infantile-onset have improved to approximately 70-80%
- Late-onset form has a more variable prognosis, with some patients having normal life expectancy with appropriate management
- Respiratory failure remains the most common cause of death in all forms
7. Diagnosis & Testing
Early and accurate diagnosis of Pompe disease is crucial for timely intervention and improved outcomes. Several diagnostic approaches are used:
Enzymatic Testing:
- Measurement of acid alpha-glucosidase (GAA) activity in blood samples:
- Dried blood spot (DBS) assay – screening test
- Lymphocyte or leukocyte assay – confirmatory test
- Cultured skin fibroblasts – gold standard but requires skin biopsy
- Results showing reduced enzyme activity (<40% of normal) suggest Pompe disease
Genetic Testing:
- Sequencing of the GAA gene to identify disease-causing mutations
- Deletion/duplication analysis if sequencing is negative
- Important for confirming diagnosis and determining carrier status
- Can help predict disease phenotype based on specific mutations
Histological Examination:
- Muscle biopsy showing glycogen accumulation in lysosomes
- Periodic acid-Schiff (PAS) staining to visualize glycogen deposits
- Electron microscopy showing abnormal lysosomes
- Less commonly used now due to availability of less invasive tests
Biomarker Testing:
- Elevated creatine kinase (CK) – marker of muscle damage
- Increased urinary glucose tetrasaccharide (Glc4)
- Elevated liver enzymes (AST, ALT)
Clinical Assessments:
- Electrocardiogram (ECG) and echocardiogram for cardiac evaluation
- Pulmonary function tests (PFTs) to assess respiratory status
- Muscle function tests to quantify weakness
- Sleep studies to evaluate for sleep-disordered breathing
Newborn Screening:
- Measurement of GAA enzyme activity in dried blood spots
- Implemented in several countries including the United States
- Allows for pre-symptomatic diagnosis and early intervention
- Has significantly improved outcomes for infantile-onset cases
The diagnostic process often follows a stepwise approach, beginning with clinical suspicion based on symptoms, followed by screening tests, and confirmation through definitive enzyme and genetic analysis. Diagnostic delays remain common in late-onset forms, with an average delay of 5-7 years from symptom onset to diagnosis.
8. Treatment Options
Treatment strategies for Pompe disease have evolved significantly over the past two decades, moving from purely supportive care to disease-modifying therapies:
Disease-Specific Treatments:
Enzyme Replacement Therapy (ERT):
- Alglucosidase alfa (Myozyme/Lumizyme) – recombinant human GAA enzyme administered intravenously
- Standard of care since FDA approval in 2006
- Typically given every two weeks as an intravenous infusion
- More effective when started early, particularly before irreversible muscle damage
- More effective for cardiac manifestations than skeletal muscle symptoms
- Clinical benefits include improved cardiac function, respiratory status, and motor development
- Limitations include variable response, development of antibodies, and high cost
Avalglucosidase alfa (Nexviazyme):
- Next-generation ERT approved in 2021
- Modified to better target the mannose-6-phosphate receptor for improved muscle uptake
- Shown to have improved efficacy compared to alglucosidase alfa in clinical trials
Chaperone Therapy:
- Small molecules that help stabilize and enhance residual enzyme activity
- Miglustat and other molecules under investigation
- Most effective for specific mutations that produce misfolded but potentially functional enzyme
Supportive Care:
Respiratory Management:
- Pulmonary function monitoring
- Non-invasive ventilation (BiPAP, CPAP)
- Invasive ventilation when necessary
- Aggressive treatment of respiratory infections
- Pulmonary rehabilitation
Cardiac Management:
- Regular cardiac monitoring
- Standard heart failure treatments when indicated
- Arrhythmia management
- Cardiac rehabilitation programs
Physical and Rehabilitation Therapy:
- Physical therapy to maintain muscle function and prevent contractures
- Occupational therapy for activities of daily living
- Speech therapy for swallowing and communication difficulties
- Assistive devices and mobility aids
Nutritional Support:
- Dietary management with specialized nutrition plans
- Swallowing evaluations and modified diets when needed
- Feeding tubes in severe cases
Emerging Treatments:
Gene Therapy:
- Adeno-associated virus (AAV) vector-mediated gene transfer
- Several approaches in clinical trials aiming to provide functional GAA gene
- Potential for long-term correction with a single treatment
Substrate Reduction Therapy:
- Reduces glycogen production to balance with impaired breakdown
- Still experimental for Pompe disease
Immunomodulation:
- Strategies to reduce antibody formation against ERT
- Protocols using rituximab, methotrexate, and immunoglobulins
Combination Therapies:
- ERT combined with chaperones for improved efficacy
- ERT with immunomodulation for immune-tolerant patients
Treatment approaches are typically multidisciplinary, involving neurologists, cardiologists, pulmonologists, metabolic specialists, physical therapists, genetic counselors, and other healthcare professionals to address the multi-system nature of the disease.
9. Prevention & Precautionary Measures
As an inherited genetic disorder, primary prevention of Pompe disease is limited to genetic counseling and reproductive options for at-risk individuals:
Genetic Counseling:
- Essential for families with history of Pompe disease
- Assessment of recurrence risk (25% for each pregnancy if both parents are carriers)
- Discussion of reproductive options
- Psychological support for affected families
Reproductive Options:
- Preimplantation genetic diagnosis (PGD) with in vitro fertilization
- Prenatal diagnosis through chorionic villus sampling (CVS) at 10-12 weeks or amniocentesis at 15-18 weeks
- Gamete donation (egg or sperm) from unaffected donors
- Adoption
Carrier Testing:
- Genetic testing to identify asymptomatic carriers
- Particularly important for individuals with family history of Pompe disease
- Cascade testing of family members once a mutation is identified
Newborn Screening:
- Early detection of affected infants before symptom onset
- Allows for prompt intervention with ERT
- Significantly improves outcomes, particularly for infantile-onset disease
- Currently implemented in many states in the US and some other countries
Secondary Prevention (preventing complications in diagnosed patients):
- Regular medical follow-up to monitor disease progression
- Prompt treatment of infections, particularly respiratory
- Vaccination against respiratory pathogens (influenza, pneumococcal)
- Proactive management of cardiac and respiratory function
- Physical therapy to maintain muscle strength and prevent contractures
- Nutritional monitoring and support
Unlike some disorders with environmental triggers, there are no known lifestyle modifications, dietary changes, or environmental precautions that can prevent the development or progression of Pompe disease. Management focuses on early diagnosis, enzyme replacement, and comprehensive care to minimize complications.
10. Global & Regional Statistics
Pompe disease affects individuals worldwide, though prevalence varies by region and ethnicity:
Global Prevalence:
- Combined prevalence of all forms: approximately 1:40,000 births
- Infantile-onset form: approximately 1:138,000 births
- Late-onset form: approximately 1:57,000 births
Regional Variations:
- African American population: approximately 1:14,000
- Dutch population: approximately 1:40,000
- Chinese population in Taiwan: approximately 1:50,000
- Australian population: approximately 1:145,000
- Portuguese population: approximately 1:600,000
Mutation Distribution:
- c.525delT – common among Dutch and European populations
- c.2560C>T (p.Arg854X) – common among African American populations
- IVS1-13T>G – common mutation in late-onset disease, accounting for about 50% of cases in Caucasians
Diagnostic Rates:
- Significant under-diagnosis, particularly of late-onset forms
- Diagnostic delays of 5-7 years common for late-onset Pompe disease
- Improved detection with expansion of newborn screening programs
Survival Rates:
- Infantile-onset (untreated): median survival 8-9 months
- Infantile-onset (treated with ERT): 5-year survival approximately 70-80%
- Late-onset: highly variable, many patients living into adulthood with treatment
- Ventilator-free survival significantly improved with ERT
Treatment Access:
- Significant disparities in access to ERT worldwide
- High cost (approximately $300,000-600,000 USD annually per patient)
- Limited availability in low and middle-income countries
- Variations in reimbursement policies and coverage
Disease Burden:
- Economic impact estimated at over $100,000 per patient per year in direct medical costs (excluding ERT)
- Significant indirect costs from lost productivity, caregiver burden
- Quality of life measures show substantial impact, particularly in advanced disease stages
The epidemiology of Pompe disease continues to evolve with improved diagnostic capabilities, expanded newborn screening, and increased awareness among healthcare providers.
11. Recent Research & Future Prospects
Research in Pompe disease has advanced rapidly in recent years, with several promising approaches under investigation:
Enhanced Enzyme Replacement Therapy:
- Avalglucosidase alfa (Nexviazyme) – approved in 2021, designed with enhanced targeting to muscle tissue
- Immune tolerance approaches to reduce antibody formation against ERT
- Alternative administration routes (subcutaneous, intrathecal) to improve distribution
- Extended half-life formulations for less frequent dosing
Gene Therapy:
- AAV-based gene therapy (SPK-3006, ACTUS-101) in clinical trials
- Liver-directed gene therapy to produce and secrete GAA enzyme
- Direct muscle-targeted gene therapy approaches
- Gene editing technologies (CRISPR/Cas9) being explored in preclinical studies
Substrate Reduction Therapy:
- Inhibition of glycogen synthase to reduce glycogen accumulation
- Combination with ERT for synergistic effects
- Several compounds in preclinical development
Autophagy Modulation:
- Targeting disrupted cellular clearance mechanisms
- Rapamycin and other mTOR inhibitors under investigation
- Approaches to enhance lysosomal function
Stem Cell Therapy:
- Hematopoietic stem cell transplantation explored in early research
- Induced pluripotent stem cells (iPSCs) for disease modeling
- Cell-based therapies for delivery of functional enzyme
RNA Therapeutics:
- Antisense oligonucleotides to correct specific splicing mutations
- mRNA therapy to deliver functional GAA enzyme instructions
Biomarkers for Disease Monitoring:
- Development of more sensitive biomarkers for disease progression
- Imaging modalities to non-invasively assess muscle glycogen content
- Patient-reported outcome measures specific to Pompe disease
Clinical Trial Landscape:
- Over 30 active clinical trials globally
- Focus on novel therapies and improved outcomes measures
- Efforts to include broader patient populations, including advanced disease
Newborn Screening Expansion:
- Development of more specific and sensitive screening methods
- Implementation in additional regions globally
- Long-term outcome studies of screened populations
The field continues to move toward personalized approaches, with treatment strategies tailored to mutation types, antibody status, and individual disease characteristics. The ultimate goal remains development of a definitive cure, though significant improvements in disease management and quality of life appear achievable in the near term.
12. Interesting Facts & Lesser-Known Insights
Pompe disease has several unique aspects that distinguish it from other rare disorders:
Historical Significance:
- First recognized lysosomal storage disorder, paving the way for understanding dozens of similar conditions
- Described during World War II under challenging circumstances
- Dr. Pompe was executed by Nazis in 1945 for resistance activities, cutting short his pioneering research
Diagnostic Journey:
- Often misdiagnosed as muscular dystrophy, polymyositis, or chronic fatigue
- Average diagnostic delay of 7+ years for late-onset forms
- Some patients see 7-10 specialists before receiving correct diagnosis
- “Limb-girdle muscular dystrophy” is a common initial misdiagnosis
Unique Clinical Features:
- Selective sparing of certain muscles (extraocular muscles rarely affected even in advanced disease)
- Diaphragm often affected earlier and more severely than other muscles
- Cognitive function typically preserved, even in infantile form
- Bassoon and French horn players have been diagnosed after experiencing specific difficulties with their instruments
Treatment Response Patterns:
- “Honeymoon period” often seen after starting ERT, followed by plateau
- Antibody development against ERT more common in infantile-onset (CRIM-negative) patients
- Individual response highly variable, even among patients with identical mutations
- Some patients develop exercise intolerance specifically during fasting or low-carbohydrate states
Pompe in Culture:
- Portrayed in the film “Extraordinary Measures” (2010) starring Harrison Ford and Brendan Fraser
- The development of Myozyme was one of the fastest orphan drug approvals in FDA history
- The patient advocacy movement significantly accelerated therapeutic development
Scientific Paradoxes:
- Despite being a monogenic disorder, there’s poor genotype-phenotype correlation for many mutations
- Secondary autophagic dysfunction may be as damaging as primary glycogen accumulation
- Exercise, typically beneficial for muscle disorders, must be carefully tailored for Pompe patients
Emerging Understanding:
- Growing recognition of non-classic manifestations including cerebral aneurysms
- Potential role of glycogen accumulation in neurons being reconsidered
- Small fiber neuropathy increasingly recognized as a component of the disease
Misconceptions:
- Despite being called a “storage” disease, it’s the disruption of cellular function rather than simple storage that causes damage
- Contrary to early belief, ERT does cross the blood-brain barrier in small amounts
- The disease is progressive but at highly variable rates; some late-onset patients remain stable for years
Pompe disease represents a remarkable story of scientific discovery, therapeutic innovation, and the power of patient advocacy in driving research for rare diseases. While challenges remain, the transformation from an invariably fatal condition to a manageable chronic disease within a single generation highlights the rapid advancement of modern medicine for rare genetic disorders.