
⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
What is Marfan Syndrome?
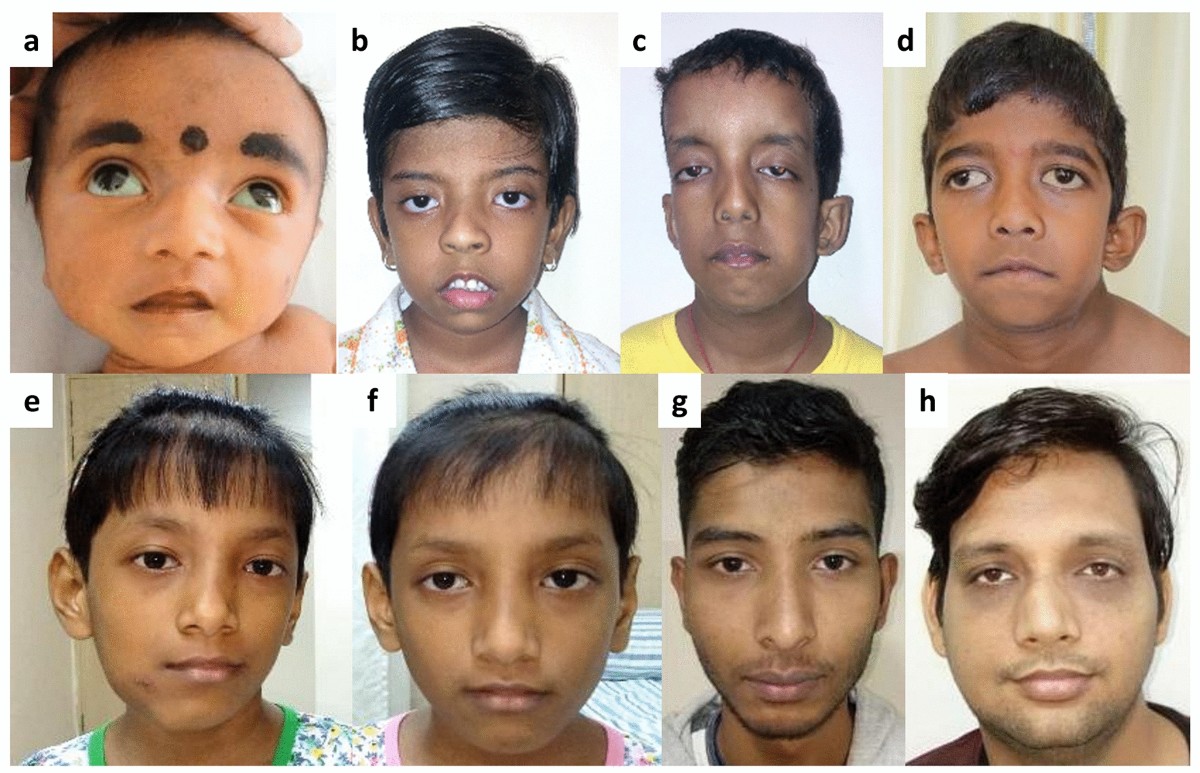
Marfan syndrome (MFS) is a systemic autosomal dominant connective tissue disorder caused by mutations in the FBN1 gene, which encodes fibrillin-1, a crucial structural protein in connective tissue microfibrils. The condition manifests as a broad phenotypic continuum ranging from mild isolated features to severe, rapidly progressive multiorgan disease.
Concise Definition
Marfan syndrome is a heritable disorder of connective tissue with a high degree of clinical variability, characterized by mutations in the fibrillin-1 gene (FBN1) on chromosome 15. The condition primarily affects the cardiovascular, skeletal, and ocular systems, with aortic root dilatation and ectopia lentis serving as cardinal clinical features.
Key Characteristics:
- Genetic basis: Autosomal dominant inheritance pattern
- Gene involved: FBN1 (fibrillin-1 gene) on chromosome 15q21.1
- Protein affected: Fibrillin-1, essential for microfibril formation
- Inheritance: 75% inherited from affected parent, 25% de novo mutations
Affected Body Parts/Organs
Primary Systems Affected:
Cardiovascular System (Most life-threatening)
- Aortic root dilatation (76% of patients)
- Aortic dissection and aneurysm
- Mitral valve prolapse (62% of patients)
- Aortic and mitral regurgitation (26% and 29% respectively)
Skeletal System
- Dolichostenomelia (disproportionately long limbs)
- Arachnodactyly (long, thin fingers and toes)
- Pectus deformity (excavatum or carinatum)
- Scoliosis and kyphosis
- Joint hypermobility
- High-arched palate
Ocular System
- Ectopia lentis (lens dislocation) – 60% of patients
- Myopia (>50% of patients)
- Increased risk of retinal detachment
- Early cataracts and glaucoma
Other Systems
- Dural ectasia (spinal cord covering enlargement)
- Spontaneous pneumothorax
- Skin striae (stretch marks)
- Central nervous system involvement (rare)
Prevalence and Significance
Global Prevalence:
- Estimated incidence: 1 in 3,000 to 1 in 5,000 individuals
- Population studies: Range from 6.5/100,000 (Denmark) to 10.2/100,000 (Taiwan)
- Gender distribution: Equal prevalence between males and females
- Ethnic distribution: No racial or ethnic predilection
- Geographic distribution: Worldwide occurrence
Clinical Significance:
- Leading genetic cause of thoracic aortic aneurysm and dissection
- Major cause of sudden cardiac death in young athletes when undiagnosed
- Substantial impact on quality of life and healthcare costs
- Improved prognosis with early detection and appropriate management
2. History & Discoveries
Timeline of Major Discoveries
1896: The Original Description French pediatrician Antoine-Bernard-Jean Marfan first described the condition in a 5.5-year-old girl named Gabrielle P., presenting her case to the Bulletin of the Medical Society of Paris. He called the condition “dolichostenomelia” (Greek for slender limbs). Ironically, modern analysis suggests Gabrielle may have had Congenital Contractural Arachnodactyly rather than what we now call Marfan syndrome.
1876: Earlier Unrecognized Description E. Williams, an ophthalmologist from Cincinnati, may have provided the first actual description of Marfan syndrome 20 years earlier, describing a brother-sister pair with ectopia lentis who were exceptionally tall and “loose-jointed.” This observation went largely unnoticed because he didn’t practice at an academic center.
Early 20th Century: Clinical Expansion
- 1902: Drs. Henri Méry and Léon Babonneix examined Gabrielle P. with new X-ray technology, initially calling her condition “hyperchondroplasia”
- 1902: Dr. Emile Achard reported similar patients and coined the term “arachnodactylie”
- 1930s-1950s: Progressive recognition of cardiovascular and ocular manifestations
Major Breakthroughs in Understanding
1950s-1960s: Cardiovascular Recognition
- Victor McKusick’s landmark work established cardiovascular manifestations as the most serious aspect
- Recognition that aortic complications were the primary cause of early death
- Mean life expectancy identified as only 32 years in 1972
1986-2010: Diagnostic Evolution
- 1986: Berlin nosology – first formal diagnostic criteria
- 1996: Ghent nosology – internationally standardized diagnostic criteria
- 2010: Revised Ghent nosology – greater emphasis on aortic root dilatation and ectopia lentis as cardinal features
1991: Genetic Breakthrough International consortium led by Dr. Anne Child identified the FBN1 gene as the causative gene for Marfan syndrome, revolutionizing diagnosis and understanding of the condition.
Modern Era Advances:
- 1994: First randomized controlled trial showing beta-blockers slow aortic root growth
- 1968: Introduction of Bentall procedure for aortic root replacement
- 2000s: Recognition of TGF-β pathway involvement in pathogenesis
- 2010s: Clinical trials of losartan as potential therapy
Evolution of Medical Understanding
Pathophysiological Evolution:
- Initial understanding: Purely structural connective tissue weakness
- Current understanding: Complex interplay involving:
- Defective fibrillin-1 leading to microfibril dysfunction
- Disrupted TGF-β signaling pathways
- Matrix metalloproteinase activation
- Inflammatory responses and tissue remodeling
Treatment Evolution:
- Pre-1970s: No effective treatment, high mortality
- 1970s-1990s: Introduction of prophylactic surgery and beta-blocker therapy
- 2000s-present: Multidisciplinary care, advanced surgical techniques, emerging medical therapies
3. Symptoms
Early Symptoms vs. Advanced-Stage Symptoms
Early/Childhood Symptoms:
Skeletal manifestations (most apparent early):
- Disproportionate height for family
- Long, thin fingers and toes (arachnodactyly)
- Chest wall deformities (pectus excavatum/carinatum)
- Scoliosis development during growth spurts
- Joint hypermobility and frequent joint pain
Ocular symptoms:
- Early myopia (nearsightedness)
- Lens dislocation (may be subtle initially)
- Visual disturbances
Cardiovascular (often asymptomatic initially):
- Heart murmurs from valve abnormalities
- Exercise intolerance (subtle early sign)
- Chest pain or palpitations
Advanced-Stage Symptoms:
Severe cardiovascular complications:
- Aortic dissection (medical emergency)
- Severe aortic or mitral regurgitation
- Heart failure symptoms
- Sudden cardiac death risk
Progressive skeletal deformities:
- Severe scoliosis requiring surgical intervention
- Protrusio acetabuli (hip socket protrusion)
- Restrictive lung disease from chest wall deformity
Advanced ocular complications:
- Retinal detachment
- Glaucoma
- Cataracts at young age
- Severe visual impairment
Common vs. Rare Symptoms
Common Symptoms (>50% of patients):
- Tall stature with arm span exceeding height
- Arachnodactyly (long, thin digits)
- Myopia
- Mitral valve prolapse
- Aortic root dilatation
- Ectopia lentis
- Skin striae not related to weight changes
- Joint hypermobility
Moderately Common Symptoms (25-50% of patients):
- Pectus deformity
- Scoliosis
- High-arched palate with dental crowding
- Dural ectasia
- Spontaneous pneumothorax
- Aortic or mitral regurgitation
Rare Symptoms (<25% of patients):
- Aortic dissection (10% lifetime risk)
- Severe heart failure
- Retinal detachment
- Central nervous system involvement
- Sleep apnea
- Hernias
Symptom Progression Over Time
Childhood and Adolescence:
- Skeletal features become more apparent during growth spurts
- Myopia typically develops and progresses
- Lens dislocation may occur
- Mild aortic root dilatation often begins
Young Adulthood (20s-30s):
- Cardiovascular manifestations become more prominent
- Peak risk period for aortic dissection begins
- Reproductive concerns arise
- Career and insurance considerations
Middle Age (40s-50s):
- Continued aortic enlargement if untreated
- Valve regurgitation may progress
- Arthritis and joint problems increase
- Secondary complications from prior surgeries
Later Life (60s+):
- Lower risk of new aortic dissection if previously managed
- Age-related complications superimposed on Marfan-related issues
- Quality of life issues from chronic pain and disability
- Potential cardiac complications from multiple prior surgeries
4. Causes
Biological Causes
Primary Genetic Cause:
- Gene: FBN1 (fibrillin-1 gene) located on chromosome 15q21.1
- Protein produced: Fibrillin-1, a 350-kDa glycoprotein
- Function: Essential component of microfibrils in connective tissue
- Mutation types: Over 1,800 different mutations identified
Molecular Pathogenesis:
Structural Deficiency: Reduced or defective fibrillin-1 production leads to:
- Weakened connective tissue structure
- Decreased elasticity and strength of tissues
- Progressive tissue failure under mechanical stress
TGF-β Dysregulation: Fibrillin-1 normally sequesters TGF-β in the extracellular matrix:
- Defective fibrillin-1 leads to excess TGF-β activity
- Results in tissue remodeling and inflammatory responses
- Contributes to progressive tissue degeneration
Matrix Metalloproteinase Activation:
- Increased MMP-2 and MMP-9 activity
- Progressive degradation of elastic fibers
- Ongoing tissue weakness and dilatation
Types of FBN1 Mutations:
- Missense mutations (60-65%): Single amino acid changes
- Nonsense mutations (10-15%): Premature stop codons
- Splice site mutations (10-15%): Affect RNA processing
- Small insertions/deletions (10-15%): Frameshift mutations
- Large deletions (rare): Removal of entire exons
Environmental Factors
Modifying Environmental Factors:
- Physical stress: High-intensity athletics may accelerate aortic dilatation
- Pregnancy: Hormonal changes and hemodynamic stress increase dissection risk
- Hypertension: Accelerates aortic enlargement
- Trauma: May precipitate aortic dissection in predisposed individuals
No Primary Environmental Causes: Marfan syndrome is purely genetic – environmental factors do not cause the condition but may influence its severity and progression.
Genetic and Hereditary Factors
Inheritance Pattern:
- Type: Autosomal dominant
- Penetrance: Extremely high (approaching 100%)
- Variable expressivity: Wide range of severity within families
Family Planning Considerations:
- Risk to offspring: 50% chance if one parent affected
- De novo mutations: Account for 25% of cases
- Paternal age effect: Advanced paternal age associated with de novo mutations
- Gonadal mosaicism: Rare but reported cause of multiple affected offspring from unaffected parents
Genetic Testing Implications:
- Detection rate: 90-95% of classical Marfan syndrome cases have identifiable FBN1 mutations
- Family screening: Essential once mutation identified in proband
- Prenatal diagnosis: Available for known familial mutations
- Preimplantation genetic diagnosis: Option for some families
Risk Triggers and Exposure Factors
Activities/Situations Increasing Risk:
- Intense physical exertion: Competitive sports, heavy lifting
- Isometric exercise: Weight lifting, excessive straining
- Valsalva maneuvers: Activities increasing thoracic pressure
- Certain medications: Fluoroquinolone antibiotics may increase dissection risk
- Stimulants: Cocaine, amphetamines, excessive caffeine
Physiological Stress Factors:
- Pregnancy: Particularly third trimester and delivery
- Infection: Severe illness may precipitate complications
- Surgery: Perioperative stress on cardiovascular system
- Sleep apnea: Increased risk of aortic complications
5. Risk Factors
Demographic Risk Factors
Age-Related Risk:
- Highest diagnostic age: Peak diagnosis occurs in adolescence and young adulthood (15-25 years)
- Aortic dissection risk: Increases with age, peak risk 30-50 years
- Childhood diagnosis: Challenging due to age-dependent penetrance of features
- Neonatal Marfan: Rare severe form with poor prognosis
Gender Considerations:
- Overall prevalence: Equal between males and females
- Aortic dissection: Males have earlier onset and higher frequency (61% of dissection cases)
- Pregnancy: Major risk factor for aortic dissection in women
- Life expectancy: Women traditionally live longer than men with Marfan syndrome
Genetic Risk Factors
Family History:
- Affected parent: 50% risk to each child
- Family history of sudden death: May indicate undiagnosed Marfan syndrome
- Connective tissue disorders in family: May suggest related conditions
Genetic Factors:
- Mutation type: Certain mutations associated with more severe phenotypes
- Exons 24-32 mutations: Associated with severe neonatal form
- Premature termination codons: Often associated with classical phenotype
- Missense mutations affecting cysteine residues: Variable severity
Occupational and Lifestyle Risk Factors
High-Risk Occupations:
- Professional athletes: Particularly in sports requiring intense cardiovascular demand
- Military personnel: Physical demands and stress
- Heavy manual labor: Risk of acute aortic complications
- Aviation: Considerations for career limitations due to cardiovascular risk
Lifestyle Factors:
- Competitive sports participation: Increases risk of sudden cardiac death
- High-intensity exercise: May accelerate aortic root growth
- Smoking: Compounds cardiovascular risks
- Poor medical compliance: Increases risk of complications
Impact of Pre-existing Conditions
Cardiovascular Conditions:
- Hypertension: Accelerates aortic root enlargement
- Bicuspid aortic valve: May compound aortic root problems
- Pregnancy: Hemodynamic changes increase dissection risk 100-fold
Other Medical Conditions:
- Sleep apnea: Associated with increased aortic root growth
- Obesity: May mask or complicate skeletal features
- Connective tissue disorders: May indicate related genetic syndromes
- Mental health conditions: Depression and anxiety common due to disease burden
Medication-Related Risks:
- Fluoroquinolone antibiotics: May increase aortic dissection risk
- Stimulant medications: Potential cardiovascular complications
- Hormonal therapies: May affect connective tissue properties
6. Complications
Cardiovascular Complications (Most Serious)
Aortic Dissection:
- Incidence: 10% of patients during lifetime
- Peak age: 30-50 years (mean 36.6 years)
- Gender distribution: 61% occur in males
- Mortality: 50% mortality rate if untreated
- Risk factors: Aortic diameter >5.0 cm, family history, pregnancy
Progressive Aortic Enlargement:
- Root dilatation: 76% of adult patients
- Annual growth rate: 0.5-1.0 mm per year typically
- Surgical threshold: Generally 5.0 cm diameter or family history of dissection at smaller size
- Complications: Aortic regurgitation, heart failure, sudden death
Valve Complications:
- Mitral valve prolapse: 62% of patients
- Mitral regurgitation: 29% of patients
- Aortic regurgitation: 26% of patients
- Progressive valve dysfunction: May require surgical intervention
Skeletal Complications
Spinal Deformities:
- Scoliosis: Progressive curvature requiring monitoring
- Kyphosis: Forward curvature of spine
- Surgical intervention: May be required for severe cases
- Restrictive lung disease: From severe chest wall deformity
Joint Problems:
- Chronic arthritis: Early onset due to joint hypermobility
- Recurrent dislocations: Particularly patella and shoulder
- Chronic pain: Significant impact on quality of life
- Functional limitation: Progressive disability in some patients
Ocular Complications
Lens Dislocation (Ectopia Lentis):
- Prevalence: 60% of patients
- Bilateral occurrence: Common
- Visual impact: Significant visual impairment
- Surgical correction: May be required after growth completion
Retinal Complications:
- Retinal detachment: Increased risk, especially with high myopia
- Glaucoma: Early onset and rapid progression
- Cataracts: Premature development
- Visual disability: May be severe and progressive
Pulmonary Complications
Spontaneous Pneumothorax:
- Incidence: 5-10% of patients
- Recurrence: High risk of repeated episodes
- Mechanism: Rupture of apical blebs
- Management: May require surgical intervention
Restrictive Lung Disease:
- Cause: Severe pectus deformity or scoliosis
- Impact: Reduced lung capacity and exercise tolerance
- Cor pulmonale: Right heart failure in severe cases
Mortality and Fatality Rates
Historical vs. Current Mortality:
- Pre-1970s: Mean life expectancy 32 years
- 1990s: Life expectancy ~45 years
- Current: Approaching normal lifespan with proper care (mid-70s)
Causes of Death:
- Aortic dissection: Leading cause (40-50% of deaths)
- Sudden cardiac death: Often first manifestation
- Heart failure: From progressive valve disease
- Surgical complications: Operative mortality 1.5-11.7% depending on urgency
Age-Specific Mortality:
- Standardized mortality ratio: 5.24 times general population
- Male mortality: 8.20 times general population
- Female mortality: 3.85 times general population
- Pediatric mortality: Higher in neonatal Marfan syndrome
Long-term Impact on Quality of Life
Physical Limitations:
- Chronic pain: 47-92% of patients report significant pain
- Exercise restrictions: Impact on career and recreational activities
- Functional disability: Progressive in some patients
- Surgical sequelae: Complications from multiple procedures
Psychosocial Impact:
- Depression and anxiety: High prevalence
- Social stigma: From physical appearance
- Career limitations: Especially affecting athletic pursuits
- Insurance discrimination: Life and health insurance challenges
- Reproductive concerns: Family planning difficulties
7. Diagnosis & Testing
Current Diagnostic Framework
2010 Revised Ghent Nosology: The gold standard for Marfan syndrome diagnosis, emphasizing aortic root dilatation and ectopia lentis as cardinal features, with genetic testing playing an important supporting role.
Cardinal Features:
- Aortic root aneurysm/dissection (Z-score ≥2 when adjusted for age and body surface area)
- Ectopia lentis (lens dislocation)
Diagnostic Criteria Rules
Rule 1: Aortic + Ectopia Lentis
- Presence of both cardinal features = definitive diagnosis
- No additional criteria needed
Rule 2: Aortic + FBN1 Mutation
- Aortic root dilatation (Z ≥ 2) + known pathogenic FBN1 mutation = diagnosis
Rule 3: Aortic + Systemic Score ≥7
- Aortic involvement + sufficient systemic features = diagnosis
- Must exclude Loeys-Dietz, Shprintzen-Goldberg, and vascular Ehlers-Danlos syndromes
Rule 4: Ectopia Lentis + FBN1 (with aortic association)
- Lens dislocation + FBN1 mutation previously associated with aortic disease = diagnosis
Rule 5: Family History Rules For patients with affected family members, diagnosis requires only one of:
- Ectopia lentis
- Systemic score ≥7
- Aortic criterion (Z ≥ 3 if <20 years; Z ≥ 2 if ≥20 years)
Systemic Score Calculation
Scoring System (points awarded):
- Wrist AND thumb signs: 3 points
- Wrist OR thumb sign: 1 point
- Pectus carinatum deformity: 2 points
- Pectus excavatum or chest asymmetry: 1 point
- Hindfoot deformity: 2 points
- Plain pes planus: 1 point
- Pneumothorax: 2 points
- Dural ectasia: 2 points
- Protrusio acetabuli: 2 points
- Reduced upper to lower segment ratio AND increased arm/height ratio AND no severe scoliosis: 1 point
- Scoliosis or thoracolumbar kyphosis: 1 point
- Reduced elbow extension: 1 point
- Facial features (3/5): 1 point
- Skin striae: 1 point
- Myopia >3 diopters: 1 point
- Mitral valve prolapse: 1 point
Total ≥7 points = positive systemic score
Essential Diagnostic Tests
Cardiovascular Assessment:
Echocardiography:
- Aortic root measurements at sinuses of Valsalva
- Assessment of valve function
- Z-score calculation based on age and body surface area
Advanced Cardiac Imaging:
- MRI or CT: For complete aortic assessment
- 3D echocardiography: Enhanced valve assessment
- Cardiac catheterization: Rarely needed
Ophthalmologic Examination:
- Slit-lamp examination: Detect ectopia lentis
- Dilated fundus examination: Assess for retinal abnormalities
- Tonometry: Screen for glaucoma
- Refraction: Document myopia severity
Skeletal Assessment:
- Physical examination: Anthropometric measurements, joint assessment
- Spine imaging: X-ray or MRI for scoliosis, dural ectasia
- Chest imaging: Document pectus deformity
- Hip imaging: Assess for protrusio acetabuli
Genetic Testing
FBN1 Gene Analysis:
- Sequencing: Complete gene sequencing to identify mutations
- Detection rate: 90-95% in classical Marfan syndrome
- Deletion/duplication analysis: For patients with negative sequencing
- Variant interpretation: Using ACMG/AMP guidelines with Marfan-specific modifications
Testing Strategy:
- Proband testing: Comprehensive FBN1 analysis
- Family screening: Targeted testing for known familial mutation
- Differential diagnosis: Testing for related genes (TGFBR1/2, COL3A1, etc.) when indicated
Genetic Counseling:
- Pre-test counseling: Discussion of implications
- Results interpretation: Clinical significance of variants
- Family planning: Reproductive options and risks
- Cascade screening: Testing of at-risk relatives
Pediatric Diagnostic Considerations
Challenges in Children:
- Age-dependent penetrance: Features may not be fully manifest
- Growth-related changes: Anthropometric measurements less reliable
- False negatives: May not meet adult criteria
Pediatric-Specific Tools:
- Kid Short Marfan (Kid-SMS) score: Risk stratification tool for children
- Modified criteria: Special considerations for patients <20 years
- Serial assessment: Regular monitoring as child grows
Differential Diagnosis
Related Connective Tissue Disorders:
- Loeys-Dietz syndrome: TGFBR1/2, SMAD3, TGFB2 mutations
- Ehlers-Danlos syndrome: Various collagen gene mutations
- Beals syndrome: FBN2 mutations
- MASS phenotype: Milder FBN1 mutations
- Marfanoid habitus: Non-syndromic tall stature
Exclusion Criteria: Must rule out discriminating features of other syndromes before diagnosing Marfan syndrome.
8. Treatment Options
Medical Management
Beta-Blocker Therapy:
- Primary medication: First-line treatment for aortic protection
- Mechanism: Reduces hemodynamic stress on aortic wall
- Agents: Atenolol, propranolol, metoprolol
- Dosing: Titrated to reduce heart rate and blood pressure
- Evidence: Proven to slow aortic root enlargement
- Initiation: Recommended when aortic dilatation present, prophylactic use in children
Angiotensin Receptor Blockers (ARBs):
- Primary agent: Losartan most studied
- Mechanism: TGF-β pathway antagonism + blood pressure reduction
- Clinical evidence:
- COMPARE study: 43% slower aortic root growth vs. no treatment
- Combination with beta-blockers shows additive benefit
- Dosing: Typically 50-100mg daily
- Patient selection: Particularly beneficial in children and young adults
Combination Therapy:
- Beta-blocker + ARB: Growing evidence for superior efficacy
- Pilot studies: Losartan added to beta-blocker therapy showed 0.10 vs 0.89 mm/year growth rate
- Safety: Generally well-tolerated combination
- Monitoring: Regular echocardiographic assessment required
Alternative Medications:
- Calcium channel blockers: For beta-blocker intolerant patients (limited evidence)
- ACE inhibitors: Theoretical benefit, limited clinical data
- Avoid: Calcium channel blockers may increase dissection risk
Surgical Management
Aortic Root Surgery:
Prophylactic Aortic Root Replacement:
- Indications:
- Aortic root diameter ≥5.0 cm
- ≥4.5 cm with family history of dissection
- Rapid enlargement (>0.5 cm/year)
- Severe aortic regurgitation
- Timing: Elective vs. emergency dramatically affects outcomes
- Mortality rates:
- Elective: 0-1.5%
- Urgent: 2.6%
- Emergency: 11.7%
- Indications:
Surgical Techniques:
Bentall procedure: Composite graft with mechanical valve
- Gold standard since 1968
- Excellent long-term results
- Requires lifelong anticoagulation
Valve-sparing operations (David or Yacoub procedures):
- Preserves native aortic valve
- Avoids anticoagulation
- Good outcomes in experienced centers
- May require valve replacement later
Ross procedure: Pulmonary autograft (rarely used in Marfan)
Outcomes of Aortic Surgery:
- Survival rates (Bentall procedure):
- 5 years: 88%
- 10 years: 81%
- 20 years: 75%
- Quality of life: Generally excellent after successful surgery
- Complications: Bleeding, infection, thromboembolism, reoperation
Other Cardiac Surgery:
- Mitral valve repair/replacement: For severe mitral regurgitation
- Mitral valve prolapse: Usually managed medically unless severe
- Arrhythmia surgery: Rarely required
Skeletal System Management
Scoliosis Treatment:
- Monitoring: Regular assessment during growth
- Bracing: For moderate curves during growth
- Surgical correction: For severe, progressive curves
- Timing: After growth completion when possible
Pectus Deformity:
- Indications for surgery: Primarily cosmetic, occasionally functional
- Techniques: Ravitch procedure, Nuss procedure
- Timing: Usually during adolescence
- Outcomes: Good cosmetic results, variable functional improvement
Ophthalmologic Management
Lens Dislocation (Ectopia Lentis):
- Conservative management: Spectacle correction when possible
- Surgical intervention:
- Lens removal with IOL implantation
- Preferably after growth completion
- Risk of retinal detachment
- Outcomes: Generally good visual rehabilitation
Other Ocular Management:
- Glaucoma: Standard medical and surgical treatment
- Cataracts: Surgical removal when vision-limiting
- Retinal detachment: Emergency surgical repair
- Regular monitoring: Annual comprehensive eye exams
Multidisciplinary Care Approach
Essential Team Members:
- Clinical geneticist: Diagnosis, genetic counseling
- Cardiologist: Cardiovascular monitoring and management
- Cardiothoracic surgeon: Aortic surgery planning and execution
- Ophthalmologist: Eye examination and treatment
- Orthopedist: Skeletal system management
- Pulmonologist: If significant pulmonary involvement
Coordination of Care:
- Regular monitoring: Annual comprehensive assessments
- Communication: Between specialists essential
- Patient education: Understanding of condition and warning signs
- Emergency planning: Recognition of aortic dissection symptoms
Emerging and Experimental Treatments
Clinical Trials:
- Losartan studies: Ongoing pediatric and adult trials
- Combination therapy: Beta-blocker + ARB studies
- Novel TGF-β antagonists: Experimental approaches
- Anti-inflammatory agents: Targeting tissue remodeling
Investigational Approaches:
- Gene therapy: CRISPR/Cas9 research in preclinical studies
- Stem cell therapy: Theoretical future applications
- Tissue engineering: Potential for aortic graft improvements
- Biomarker development: For monitoring disease progression
9. Prevention & Precautionary Measures
Primary Prevention (Genetic)
Genetic Counseling:
- Pre-conception counseling: Risk assessment and reproductive options
- Inheritance pattern: 50% risk to offspring from affected parent
- Reproductive options:
- Natural conception: With prenatal diagnosis
- Preimplantation genetic diagnosis (PGD): IVF with genetic testing
- Donor gametes: To avoid transmission
- Adoption: Alternative family building
Prenatal Diagnosis:
- Availability: For known familial mutations
- Timing: Chorionic villus sampling (10-12 weeks) or amniocentesis (15-18 weeks)
- Accuracy: >99% for known mutations
- Considerations: Pregnancy itself carries increased risk for women with Marfan
Cascade Family Screening:
- Family assessment: All first-degree relatives should be evaluated
- Genetic testing: For known familial mutations
- Clinical screening: Even without genetic testing available
- Early identification: Enables preventive management
Secondary Prevention (Early Detection and Management)
Regular Surveillance:
Cardiovascular Monitoring:
- Frequency:
- Children: Every 6-12 months
- Adults with stable aorta <4.5cm: Annually
- Adults with enlarging aorta: Every 6 months
- After aortic surgery: Annually
- Imaging: Echocardiography primary, MRI/CT for complete assessment
- Parameters: Aortic root dimensions, valve function, other cardiac structures
Ophthalmologic Surveillance:
- Frequency: Annual comprehensive eye exams
- Focus: Lens position, retinal status, intraocular pressure
- High myopia: May require more frequent monitoring
- Family history: Of retinal detachment warrants closer follow-up
Skeletal Monitoring:
- Growth periods: More frequent assessment during rapid growth
- Scoliosis screening: Regular clinical and radiographic assessment
- Adult monitoring: For progressive arthritis and joint problems
Lifestyle Modifications and Precautions
Exercise Guidelines:
Recommended activities:
- Low to moderate intensity exercise (4-6 METs)
- Walking, swimming, cycling
- Skill-based sports rather than intense competition
Restricted activities:
- Contact sports (football, hockey, martial arts)
- Competitive athletics to exhaustion
- Isometric exercises (weight lifting, push-ups, sit-ups)
- Activities with risk of bodily collision
Individual assessment: Based on aortic dimensions and cardiac function
Pregnancy Considerations:
- Pre-pregnancy counseling: Cardiac risk assessment essential
- High-risk pregnancy: Requires specialized maternal-fetal medicine care
- Aortic diameter limits: Generally <4.0 cm considered safer for pregnancy
- Delivery planning: Mode of delivery based on aortic status
- Postpartum monitoring: Risk continues for several weeks after delivery
Occupational Considerations:
- Career guidance: Early counseling about appropriate career paths
- Physical demands: Avoid careers requiring intense physical exertion
- Medical clearance: May be required for certain occupations
- Insurance implications: Potential limitations in life and disability insurance
Environmental and Situational Precautions
Medication Precautions:
- Avoid fluoroquinolone antibiotics: May increase aortic dissection risk
- Stimulant medications: Use with caution due to cardiovascular effects
- Decongestants: Avoid excessive use due to cardiovascular stimulation
- Careful anesthesia: Hemodynamic monitoring during procedures
Situational Awareness:
- Air travel: Generally safe, but ensure access to medical care
- High altitude: Potential increased cardiac stress
- Hot weather: Maintain adequate hydration
- Stress management: Chronic stress may worsen cardiovascular risks
Emergency Preparedness:
- Warning signs education: Recognition of aortic dissection symptoms
- Emergency contacts: Immediate access to specialized care
- Medical identification: Bracelet or card identifying condition
- Emergency action plan: For family members and caregivers
Community and Population-Level Prevention
Awareness Programs:
- Professional education: Training for healthcare providers
- Public awareness: Recognition of signs and symptoms
- Screening programs: In high-risk populations
- Research participation: Contributing to improved understanding
Healthcare System Improvements:
- Multidisciplinary clinics: Coordinated care models
- Genetic services: Access to genetic counseling and testing
- Emergency protocols: Rapid recognition and treatment of complications
- Quality measures: Monitoring outcomes and improving care
10. Global & Regional Statistics
Worldwide Prevalence and Incidence
Global Estimates:
- Traditional estimate: 1 in 5,000 individuals (widely cited but original source unclear)
- Modern population studies: Range from 1 in 3,000 to 1 in 10,000
- Geographic variation: No significant differences between regions or ethnicities
- Gender distribution: Equal prevalence between males and females
Regional Population Studies:
Denmark (Comprehensive National Study):
- Study period: 1977-2014
- Point prevalence: 6.5 per 100,000 (2014)
- Incidence: 0.19 per 100,000 annually
- Methodology: Rigorous application of 2010 Ghent criteria
- Total confirmed cases: 412 patients
Taiwan (National Health Insurance Database):
- Study period: 2000-2012
- Prevalence: 10.2 per 100,000 (95% CI: 9.8-10.7)
- Total patients: 2,329 identified
- Gender distribution: 58% male
- Peak diagnosis age: 15-19, 10-14, and 20-24 years
Hong Kong (Hospital-Based Study):
- Study period: 1997-2006
- Hospital admissions: 525 patients with Marfan syndrome
- Gender distribution: 310 male, 215 female
- Mean age at admission: 19.8 years (males), 18.7 years (females)
Norway (Population Cohort):
- Study design: Follow-up of 84 adults with Marfan syndrome
- Follow-up period: 2003-2015
- Mortality assessment: Standardized mortality ratios calculated
- Long-term outcomes: Survival and cardiovascular events analyzed
Mortality and Survival Statistics
Historical vs. Current Life Expectancy:
- 1972: Mean age at death 32 years (Murdoch et al.)
- 1995: Life expectancy improved to ~45 years
- 2009: Further improvement to mid-70s with modern care
- Current: Approaching normal lifespan with optimal management
Mortality Ratios (Norwegian Study):
- Overall standardized mortality ratio: 5.24 (95% CI: 3.00-8.51)
- Male mortality ratio: 8.20 (95% CI: 3.54-16.16)
- Female mortality ratio: 3.85 (95% CI: 1.66-7.58)
- Interpretation: 5-8 times higher mortality risk than general population
Age-Specific Mortality Patterns:
- Taiwan study findings:
- Average annual mortality: 0.23% (69 deaths over study period)
- Cardiac causes: 58% of deaths (40 patients with dissection/sudden death)
- More than half of deaths and dissections occurred before age 40
Aortic Dissection Statistics:
- Lifetime incidence: 10% of patients (Taiwan study: 226/2,329 patients)
- Gender distribution: 61% occur in males
- Mean age at dissection: 36.6 ± 10.7 years
- Age-specific probability of freedom from dissection:
- Age 20: 99%
- Age 40: 80%
- Age 50: 66%
Cardiovascular Intervention Outcomes
Surgical Statistics (Taiwan Study):
- Total interventions: 360 patients underwent cardiovascular procedures
- Operative mortality:
- Elective procedures: 0%
- Emergency procedures: 8%
- Interpretation: Emergency surgery carries significantly higher risk
Johns Hopkins Experience (Historical Data):
- Aortic root replacements: 231 patients (1976-1997)
- 30-day mortality:
- Elective surgery: 0% (198 patients)
- Urgent surgery: 6% (2/33 patients)
- Long-term survival:
- 5 years: 88%
- 10 years: 81%
- 20 years: 75%
Regional Healthcare Outcomes
Quality of Care Indicators:
- Diagnostic delay: Variable worldwide, improving with awareness
- Access to genetic testing: Limited in resource-constrained settings
- Specialized care availability: Concentrated in academic medical centers
- Multidisciplinary clinics: Becoming standard in developed countries
Healthcare System Performance:
- Developed countries: Generally good outcomes with organized care
- Developing countries: Limited data, presumed higher mortality
- Rural vs. urban: Significant disparities in access to specialized care
- Insurance coverage: Variable impact on treatment access
Demographic Trends and Projections
Age at Diagnosis Patterns:
- Peak diagnosis ages:
- Adolescence (15-19 years): Growth spurt reveals features
- Young adulthood (20-24 years): Cardiovascular complications
- Childhood (10-14 years): Family screening
- Diagnostic challenges:
- Pediatric cases: Features may not be fully manifest
- Adult diagnosis: May represent milder forms or late recognition
Population Growth Projections:
- Danish predictions: Prevalence expected to increase 0.17 patients/100,000 annually
- Factors influencing growth:
- Improved survival leading to larger living population
- Better diagnostic recognition
- Genetic counseling affecting reproductive decisions
- Mortality impact: If relative mortality risk <2.0, population will continue expanding
Economic Burden
Healthcare Costs:
- Comprehensive cost analysis: Limited published data
- Major cost drivers:
- Cardiac surgery (highest single cost)
- Lifelong monitoring and follow-up
- Emergency care for complications
- Genetic testing and counseling
- Quality of life impact: Significant but improving with better treatment
Societal Impact:
- Work productivity: Limited by activity restrictions and health issues
- Educational impact: May affect career choices and opportunities
- Family burden: Genetic counseling and cascade screening costs
- Insurance implications: Life and health insurance considerations
11. Recent Research & Future Prospects
Gene Therapy and CRISPR Technology
CRISPR/Cas9 Applications for Marfan Syndrome:
Current Research Status:
- Proof of concept: Successfully demonstrated in laboratory studies
- Target gene: FBN1 mutations being corrected using CRISPR/Cas9
- Research achievements:
- T7498C mutation successfully corrected in human embryos (2017)
- MYBPC3 deletion corrected in cardiac applications
- Mouse models showing improved phenotypes after gene editing
Technical Challenges:
- Delivery systems: Getting CRISPR tools to all affected tissues
- Timing considerations: Optimal age for intervention (embryonic vs. postnatal)
- Target tissue selection: Which cells/organs should be prioritized
- Off-target effects: Ensuring precise editing without unintended consequences
Future Potential:
- Definitive maybe: Expert consensus on eventual applicability
- Timeline: Several years away from human clinical trials
- Complementary approaches: May work alongside traditional treatments
- Regulatory pathway: Will require extensive safety and efficacy studies
Advanced Pharmacological Research
TGF-β Pathway Modulation:
- Mechanism: Targeting the fundamental pathophysiology
- Current agents: Losartan showing promise in clinical trials
- Novel compounds: Development of more specific TGF-β antagonists
- Combination approaches: Multiple pathway inhibition strategies
Matrix Metalloproteinase Inhibitors:
- Rationale: Preventing elastic fiber degradation
- Experimental agents: Various MMP inhibitors under investigation
- Challenges: Specificity and avoiding side effects
- Potential: Slowing or preventing tissue deterioration
Anti-inflammatory Strategies:
- Targeting: Chronic inflammatory component of tissue remodeling
- Agents under study: Various anti-inflammatory compounds
- Integration: With existing cardiovascular protective strategies
Biomarker Development
Circulating Biomarkers:
- Purpose: Monitoring disease progression and treatment response
- Candidates:
- Matrix breakdown products
- Inflammatory markers
- Growth factors and cytokines
- Clinical utility: Reducing need for frequent imaging
Genetic Biomarkers:
- Variant reclassification: Ongoing improvement in mutation interpretation
- Modifier genes: Identifying factors that influence disease severity
- Pharmacogenomics: Predicting treatment response based on genetics
Cardiovascular Research Advances
Surgical Innovations:
- Valve-sparing techniques: Continued refinement and improvement
- Minimally invasive approaches: Reducing surgical morbidity
- Tissue engineering: Development of improved graft materials
- Robotic surgery: Potential applications in cardiac procedures
Medical Management Optimization:
- Combination therapy trials: Beta-blockers plus ARBs
- Personalized dosing: Based on individual patient characteristics
- Pediatric applications: Age-specific treatment protocols
- Long-term safety: Extended follow-up of treatment regimens
Diagnostic Technology Improvements
Imaging Advances:
- 4D Flow MRI: Better assessment of aortic hemodynamics
- Artificial intelligence: Automated measurement and interpretation
- Portable ultrasound: Improving access to monitoring
- Multi-parametric imaging: Comprehensive tissue characterization
Genetic Testing Evolution:
- Whole genome sequencing: Identifying novel disease genes
- RNA analysis: Understanding splicing variants
- Functional studies: Confirming pathogenicity of variants
- Cost reduction: Making genetic testing more accessible globally
Regenerative Medicine Research
Stem Cell Applications:
- Cardiovascular repair: Potential for treating valve disease
- Connective tissue regeneration: Theoretical applications
- iPSC models: Disease modeling and drug screening
- Clinical trials: Early-stage investigations
Tissue Engineering:
- Artificial blood vessels: Improved materials for aortic replacement
- Valve reconstruction: Bioengineered valve components
- Scaffold technology: Supporting tissue regeneration
- 3D printing: Custom prosthetic development
International Collaborative Research
Global Research Networks:
- GenTAC Alliance: NIH-funded research consortium
- Marfan Foundation Research: Supporting innovative studies
- International collaborations: Multi-country research initiatives
- Patient registries: Large-scale data collection efforts
Current Major Studies:
- Pediatric Heart Network: Losartan vs. atenolol trials
- European studies: COMPARE trial and follow-up studies
- Genetic consortiums: Variant curation and interpretation
- Natural history studies: Long-term outcome tracking
Future Therapeutic Horizons (5-10 Years)
Near-term Prospects (2025-2030):
- Combination medical therapy: Beta-blockers + ARBs becoming standard
- Improved surgical techniques: Better outcomes and lower morbidity
- Enhanced genetic testing: More comprehensive and accessible
- Personalized medicine: Treatment based on individual genetic profiles
Medium-term Prospects (2030-2035):
- Gene therapy trials: First human clinical studies
- Novel pharmacological agents: Next-generation TGF-β modulators
- Advanced biomarkers: Clinical integration for monitoring
- Regenerative approaches: Tissue engineering applications
Long-term Vision (2035+):
- Curative gene therapy: Potential for definitive treatment
- Prevention strategies: Stopping disease before symptoms appear
- Artificial organs: Advanced prosthetic replacements
- Complete disease modification: Changing natural history fundamentally
Research Funding and Priorities
Major Funding Sources:
- National Institutes of Health: Significant federal investment
- Marfan Foundation: Private foundation research support
- International organizations: Global collaborative funding
- Pharmaceutical industry: Drug development partnerships
Priority Research Areas:
- Gene therapy development: Moving toward clinical applications
- Pharmacological innovation: Novel therapeutic targets
- Surgical technique advancement: Improving outcomes and reducing risks
- Biomarker validation: Clinical utility demonstration
- Quality of life research: Addressing patient-reported outcomes
12. Interesting Facts & Lesser-Known Insights
Historical Mysteries and Medical Detective Work
The Abraham Lincoln Controversy: One of medicine’s most enduring debates centers on whether America’s 16th president had Marfan syndrome. The theory was first proposed in 1962 by Dr. A.M. Gordon, based on Lincoln’s remarkable stature—6’4″ with extraordinarily long limbs and large hands and feet. Supporting evidence includes his mother’s similar tall, lanky build and a descendant of Lincoln’s great-great-grandfather who was diagnosed with Marfan syndrome.
However, Lincoln lacked key Marfan features: he was never known to have heart murmurs, eye problems, or joint hypermobility. In fact, he remained physically strong throughout his life, able to chop wood just weeks before his assassination. Some experts now suggest he may have had Multiple Endocrine Neoplasia 2B (MEN2B), a different genetic condition that could have eventually caused cancer. The debate continues, with DNA testing attempts yielding inconclusive results.
The Original Patient’s Ironic Diagnosis: In a fascinating medical irony, the 5.5-year-old girl “Gabrielle P.” whom Antoine Marfan described in 1896—and after whom the syndrome was named—likely didn’t actually have Marfan syndrome. Modern analysis of Marfan’s original case description suggests Gabrielle probably had Congenital Contractural Arachnodactyly (CCA), a related but distinct genetic condition. The true first description may have been made 20 years earlier by American ophthalmologist E. Williams, whose work went largely unnoticed.
Famous Figures and Marfan Syndrome
Musical Virtuosos: Several legendary musicians likely had Marfan syndrome, which may have contributed to their extraordinary abilities:
Niccolò Paganini (1782-1840): The legendary violinist’s incredibly long, flexible fingers allowed him to play techniques considered impossible by others. His pale complexion, thin frame, and hypermobile joints all suggest Marfan syndrome. Audiences were so amazed by his abilities that rumors spread he had made a pact with the devil.
Sergei Rachmaninoff (1873-1943): The composer’s enormous hands (he could span 12 piano keys) and tall, thin stature suggest possible Marfan syndrome. His hand span was so unusual that many pianists today cannot perform his compositions as originally written.
Robert Johnson (1911-1938): The legendary blues musician’s tall, thin frame and long fingers may have contributed to his distinctive guitar technique. His sudden death at age 27 could have been from undiagnosed cardiovascular complications.
Contemporary Figures:
Joey Ramone (1951-2001): The Ramones lead singer, standing 6’6″ with his characteristic hunched posture, was open about having Marfan syndrome. He became an spokesperson for The Marfan Foundation.
Jonathan Larson (1960-1996): The composer of the musical “Rent” died suddenly from aortic dissection just before his show’s opening night. His death brought significant attention to Marfan syndrome and the importance of early diagnosis.
Surprising Medical Connections
The Sports Medicine Revolution: Marfan syndrome research has fundamentally changed sports medicine. The condition led to the development of comprehensive cardiac screening for athletes and established the principle that tall, thin athletes require special cardiovascular evaluation. This has saved countless lives by identifying undiagnosed cases before sudden cardiac death occurs.
The TGF-β Discovery: Research into Marfan syndrome pathogenesis revealed the crucial role of TGF-β (transforming growth factor-beta) signaling in connective tissue disorders. This discovery has had far-reaching implications beyond Marfan syndrome, contributing to understanding of cancer, wound healing, and aging processes.
Myths vs. Medical Facts
Myth: “Marfan syndrome patients are always extremely tall” Fact: While most patients are taller than average for their family, height alone is not diagnostic. Some patients have normal or even below-average height. The diagnosis depends on multiple criteria, not just stature.
Myth: “People with Marfan syndrome can’t exercise at all” Fact: Most patients can engage in regular, moderate exercise. Restrictions apply to high-intensity competitive sports and activities with risk of bodily collision. Swimming, walking, and skill-based activities are typically encouraged.
Myth: “Marfan syndrome always causes heart problems” Fact: While cardiovascular involvement is common and serious, some patients have minimal cardiac involvement. Regular monitoring is essential because problems can develop over time.
Myth: “All tall, thin people should be tested for Marfan syndrome” Fact: Most tall, thin individuals don’t have Marfan syndrome. The condition requires multiple specific criteria for diagnosis, including family history, genetic testing, or characteristic complications.
Fascinating Biological Insights
The Connective Tissue Paradox: In Marfan syndrome, tissues are simultaneously too weak (leading to aortic enlargement) and too stiff (causing joint problems). This paradox occurs because fibrillin-1 deficiency affects different tissue properties in different ways—some lose elasticity while others become hyperextensible.
The Growth Factor Connection: Fibrillin-1 acts as a “molecular glue” that holds growth factors in place. When it’s defective, growth factors like TGF-β are released inappropriately, causing a cascade of tissue changes. This discovery revolutionized understanding of how genetic mutations can have widespread effects.
The Evolutionary Perspective: Some researchers theorize that Marfan-related genes may have provided evolutionary advantages in certain environments. The increased height and limb length could have been beneficial for reaching food sources or in other survival scenarios, explaining why these mutations persist in the population.
Cultural and Social Impact
The “Marfanoid” Aesthetic: The tall, thin body type associated with Marfan syndrome has sometimes been idealized in certain cultures and fashion industries. However, this has led to problematic associations and misunderstanding of the medical condition underlying these physical characteristics.
Medical Terminology Evolution: The term “arachnodactyly” (spider-like fingers) was coined specifically to describe the long, thin digits seen in Marfan syndrome. This term has since been adopted to describe similar findings in other genetic conditions.
Insurance and Discrimination: Historically, people with Marfan syndrome faced significant discrimination in life insurance and employment. Legal protections have improved, but challenges remain. Some countries now prohibit genetic discrimination in insurance.
Technological Advances Inspired by Marfan Research
Imaging Innovations: The need to precisely measure aortic dimensions in Marfan patients has driven advances in cardiac imaging, including 3D echocardiography and cardiac MRI techniques. These innovations benefit all cardiac patients.
Genetic Testing Evolution: Marfan syndrome was among the first conditions to demonstrate the clinical utility of genetic testing for inherited diseases. The experience gained has informed approaches to many other genetic conditions.
Surgical Technique Development: The high risk of aortic complications in young Marfan patients motivated development of improved surgical techniques, including valve-sparing operations that preserve the patient’s own heart valve while replacing the diseased aortic root.
Unexpected Research Applications
Aging Research: Studies of connective tissue changes in Marfan syndrome have provided insights into normal aging processes, particularly how extracellular matrix changes contribute to age-related tissue deterioration.
Cancer Research: The TGF-β pathway disruption seen in Marfan syndrome has informed cancer research, as this same pathway is often dysregulated in various cancers.
Wound Healing: Understanding how fibrillin-1 affects tissue repair has led to research into improved wound healing strategies and tissue engineering applications.
Modern Mysteries Still Being Solved
The Neonatal Marfan Enigma: Severe neonatal Marfan syndrome remains poorly understood. Why some mutations cause devastating early-onset disease while others cause mild adult symptoms is still being investigated.
The Penetrance Puzzle: Why some individuals with pathogenic FBN1 mutations have minimal symptoms while others develop severe, life-threatening complications remains an active area of research. Modifier genes and environmental factors likely play important roles.
The Athletic Advantage Question: Some researchers question whether certain Marfan-related traits might actually provide advantages in specific sports, creating an ethical dilemma about participation restrictions.
Looking to the Future
The Personalized Medicine Revolution: Marfan syndrome research is at the forefront of personalized medicine, with treatment recommendations increasingly based on individual genetic profiles, family history, and specific mutation characteristics.
The Gene Therapy Frontier: As one of the most well-characterized single-gene disorders, Marfan syndrome represents an ideal target for gene therapy approaches. Early research suggests this could eventually provide curative treatment.
The Quality of Life Focus: Modern Marfan syndrome research increasingly emphasizes patient-reported outcomes and quality of life measures, recognizing that survival is no longer the only important endpoint for most patients.
Conclusion
Marfan syndrome stands as a testament to the power of medical research and multidisciplinary care. From Antoine Marfan’s original description in 1896 to today’s sophisticated genetic and therapeutic approaches, the journey has been remarkable. What was once a condition with a mean life expectancy of 32 years has been transformed into a manageable chronic disease with near-normal lifespan for many patients.
The story of Marfan syndrome illustrates several important medical principles: the value of genetic research, the importance of early diagnosis, the power of preventive medicine, and the necessity of multidisciplinary care. It also demonstrates how understanding one rare disease can have far-reaching implications for medicine as a whole—from TGF-β pathway research that benefits cancer patients to surgical techniques that improve outcomes for all cardiac patients.
Looking forward, the prospects are encouraging. Gene therapy approaches offer the potential for curative treatment, while continued pharmacological research may provide better medical management options. Improved diagnostic techniques and global awareness programs promise earlier detection and better outcomes worldwide.
Perhaps most importantly, the Marfan syndrome community—patients, families, researchers, and healthcare providers—exemplifies how collaboration can accelerate progress. The Marfan Foundation and similar organizations worldwide continue to drive research, support families, and advocate for improved care.
For individuals living with Marfan syndrome today, the message is one of hope. With proper medical care, appropriate lifestyle modifications, and regular monitoring, most people with Marfan syndrome can expect to live long, productive lives. The key is early diagnosis, comprehensive care, and staying connected with the medical advances that continue to improve outcomes.
The journey from a mysterious connective tissue disorder to a well-understood genetic condition with effective treatments represents one of medicine’s great success stories. As research continues and new therapies emerge, the future for people with Marfan syndrome has never been brighter.