
⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
What is Hypophosphatasia?
Hypophosphatasia (HPP) is a rare, inherited metabolic bone disease characterized by defective mineralization of bones and teeth. The condition is caused by mutations in the ALPL gene, which encodes the tissue-nonspecific alkaline phosphatase enzyme (TNSALP). This enzyme plays a crucial role in the mineralization process by breaking down certain compounds that inhibit the formation of hydroxyapatite crystals, the mineral component of bones and teeth.
When TNSALP activity is reduced, these inhibitory compounds—particularly inorganic pyrophosphate (PPi), pyridoxal 5′-phosphate (PLP, a form of vitamin B6), and phosphoethanolamine (PEA)—accumulate in the extracellular space, preventing normal mineralization. This leads to various skeletal abnormalities and systemic complications.
Affected Body Parts/Organs
Hypophosphatasia primarily affects the skeletal system and teeth but can impact multiple organ systems depending on the severity:
Skeletal System:
- Bones (particularly the long bones, ribs, and skull)
- Growth plates
- Joints
Dental Structures:
- Teeth (premature loss of primary teeth, dental caries)
- Alveolar bone (supporting structure for teeth)
Respiratory System:
- Chest deformities leading to respiratory insufficiency
- Poorly mineralized ribs causing respiratory failure in severe infantile cases
Neurological System:
- Brain (increased intracranial pressure due to craniosynostosis)
- Spinal cord (complications from vertebral fractures)
- Peripheral nerves (in some cases)
Renal System:
- Kidneys (nephrocalcinosis in some cases)
Musculoskeletal System:
- Muscles (weakness, pain)
- Tendons and ligaments (calcification leading to inflammation)
Prevalence and Significance
Hypophosphatasia is considered a rare disease with variable prevalence across different populations:
- Overall incidence: Approximately 1 in 100,000 live births for severe forms
- Mild forms: Likely underdiagnosed, with estimates suggesting 1 in 6,370 to 1 in 2,500 prevalence
- Geographic variations:
- Higher prevalence in certain populations:
- 1 in 2,500 in Manitoba Mennonites of Canada
- 1 in 6,370 in Europeans
- Rarer in African and Asian populations
- Higher prevalence in certain populations:
The significance of HPP stems from its:
Clinical Severity Spectrum: Ranges from death in utero to isolated dental issues in adulthood, with six recognized clinical forms (perinatal lethal, perinatal benign, infantile, childhood, adult, and odontohypophosphatasia)
Mortality Rate: Nearly 100% in the perinatal lethal form without treatment; approximately 50% in the infantile form
Morbidity: Significant impact on quality of life due to:
- Chronic bone pain
- Recurrent fractures
- Impaired mobility
- Dental complications
- Growth failure in children
Diagnostic Challenges: Often misdiagnosed as more common conditions, delaying proper treatment
Treatment Advances: Development of enzyme replacement therapy (asfotase alfa) has dramatically improved outcomes for severe forms, transforming previously fatal conditions into manageable diseases
Economic Impact: Significant healthcare costs associated with managing complications, especially before the advent of targeted therapy
Genetic Implications: Autosomal recessive inheritance in severe forms and autosomal dominant in milder forms, with implications for family planning
Hypophosphatasia represents a perfect example of how understanding the molecular basis of a disease can lead to targeted therapies that transform outcomes. This makes it an important model in the field of rare metabolic bone disorders and enzyme replacement therapies.
2. History & Discoveries
First Identification
Hypophosphatasia was first identified and described in 1948 by Dr. John Campbell Rathbun, a Canadian pediatrician at the University of Toronto. Rathbun published a case report in the American Journal of Diseases of Children entitled “Hypophosphatasia: A New Developmental Anomaly.” The paper described six cases of infants with a previously unrecognized skeletal disorder characterized by:
- Defective skeletal mineralization
- Increased urinary excretion of phosphoethanolamine
- Profoundly decreased alkaline phosphatase activity in serum and tissues
Rathbun astutely recognized that the condition was different from other known forms of rickets and named it “hypophosphatasia” to reflect the low alkaline phosphatase activity that distinguished it from other metabolic bone diseases, which often featured elevated alkaline phosphatase levels.
Key Historical Figures and Discoveries
The understanding of hypophosphatasia has evolved through several key discoveries:
1948: John Campbell Rathbun – First clinical description and naming of the condition
1953: Schlesinger et al. – Established the hereditary nature of the disease and described the autosomal recessive inheritance pattern in severe cases
1957: Fraser – Described the less severe childhood form of hypophosphatasia, expanding the clinical spectrum
1970: Whyte et al. – Characterized the biochemical abnormalities, particularly the elevated substrate levels of TNSALP in bodily fluids
1988: Weiss et al. – Identified the human TNSALP gene (ALPL) located on chromosome 1p36.1-34
1988-1992: Multiple researchers – Identification of the first pathogenic mutations in the ALPL gene
1990s: Henthorn, Millán, and colleagues – Detailed characterization of the structure and function of TNSALP enzyme
2000: Whyte et al. – Established the natural history of the various forms of hypophosphatasia
2008: Millán et al. – Development of the first mouse model of hypophosphatasia and experimental enzyme replacement therapy
2012: Whyte et al. – First successful clinical trials of asfotase alfa (enzyme replacement therapy)
2015: FDA and EMA approval of asfotase alfa (Strensiq®) – First approved therapy for hypophosphatasia
Major Breakthroughs in Research and Treatment
Several key breakthroughs have transformed our understanding and management of hypophosphatasia:
Biochemical Characterization (1950s-1970s):
- Identification of elevated phosphoethanolamine, pyridoxal 5′-phosphate, and inorganic pyrophosphate in HPP patients
- Understanding the role of these substrates in inhibiting mineralization
Genetic Discoveries (1980s-1990s):
- Cloning of the ALPL gene
- Identification of hundreds of different mutations
- Recognition of genotype-phenotype correlations
- Understanding dominant-negative effects in milder forms
Pathophysiological Insights (1990s-2000s):
- Clarification of TNSALP’s role in hydroxyapatite crystal formation
- Understanding the function of TNSALP in vitamin B6 metabolism and its relation to seizures in severe HPP
- Recognition of the importance of the TNSALP-PPi-ANKH axis in mineralization
Therapeutic Development (2000s-2015):
- Creation of mouse models of HPP
- Development of bone-targeted recombinant enzyme replacement therapy
- Successful clinical trials showing dramatic improvements in survival and bone mineralization
- FDA approval of asfotase alfa in 2015, the first specific treatment for HPP
Clinical Management Advances (2010s-present):
- Development of multidisciplinary management approaches
- Improved dental treatments
- Better understanding of pain management in HPP
- Recognition and management of non-skeletal manifestations
Evolution of Medical Understanding
The conceptualization of hypophosphatasia has evolved significantly over time:
Initial Recognition Phase (1940s-1950s):
- HPP identified as a distinct clinical entity
- Recognition of its hereditary nature
- Distinction from other forms of rickets and osteomalacia
Biochemical Characterization Phase (1950s-1970s):
- Understanding the relationship between alkaline phosphatase deficiency and abnormal mineralization
- Recognition of the role of substrate accumulation
- Development of biochemical diagnostic criteria
Genetic Era (1980s-2000s):
- Identification of the ALPL gene
- Recognition of the heterogeneity of mutations
- Understanding of inheritance patterns
- Appreciation of the spectrum of clinical severity
Therapeutic Revolution (2000s-present):
- Shift from supportive care to disease-modifying treatment
- Profound improvement in outcomes, particularly for severe forms
- Recognition of HPP as a treatable condition rather than a fatal one
Comprehensive Management Phase (2010s-present):
- Development of interdisciplinary care approaches
- Recognition of the importance of early diagnosis
- Understanding of HPP as a lifelong condition requiring ongoing management
- Recognition of atypical presentations and previously undiagnosed milder forms
The journey from Rathbun’s initial description to the current comprehensive understanding of hypophosphatasia exemplifies how advances in genetics, biochemistry, and molecular medicine can transform rare disease management. The development of effective enzyme replacement therapy for HPP represents one of the most significant successes in the treatment of rare metabolic bone disorders, turning what was once a frequently fatal condition into a manageable, chronic disease.
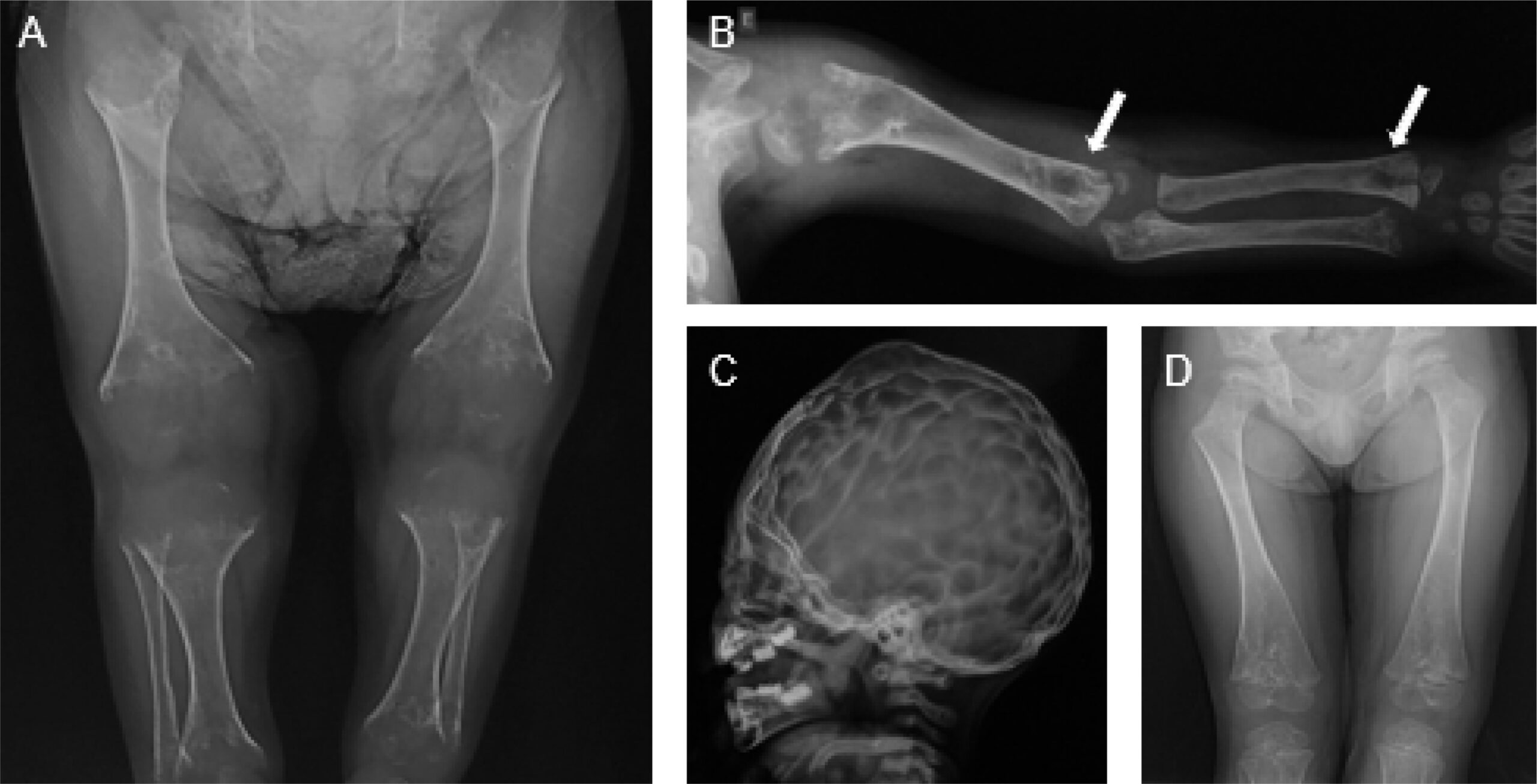
3. Symptoms
Clinical Forms and Manifestations
Hypophosphatasia presents across a spectrum of severity, conventionally classified into six forms based on age at onset and symptom severity:
- Perinatal Lethal Form
- Perinatal Benign Form
- Infantile Form
- Childhood Form
- Adult Form
- Odontohypophosphatasia
Each form has distinctive presentations and progression patterns.
Early Symptoms by Clinical Form
Perinatal Lethal Form (evident prenatally or at birth):
- Profound skeletal hypomineralization visible on prenatal ultrasound
- Shortened and deformed limbs
- Caput membranaceum (soft skull with minimal mineralization)
- Skin-covered osteochondral spurs protruding from forearms or legs
- Respiratory insufficiency due to poorly mineralized chest and lung hypoplasia
- Hypercalcemia and hyperphosphatemia
- Seizures (vitamin B6-dependent)
Perinatal Benign Form:
- Skeletal abnormalities detected prenatally
- Better mineralized skeleton than the lethal form
- Limb bowing
- Short ribs
- Spontaneous improvement after birth (hallmark feature)
Infantile Form (onset <6 months):
- Failure to thrive
- Hypotonia (poor muscle tone)
- Delayed motor milestones
- Rachitic chest deformities (including Harrison’s groove)
- Craniosynostosis (premature fusion of skull sutures)
- Raised intracranial pressure
- Hypercalcemia and hypercalciuria
- Respiratory difficulties
- Poor feeding
- Irritability
- Vitamin B6-responsive seizures
- Blue sclerae
Childhood Form (onset 6 months to 18 years):
- Premature loss of primary teeth (especially incisors) with intact roots
- Short stature
- Delayed walking
- Waddling gait
- Bone pain
- Recurrent fractures
- Craniosynostosis (less common than in infantile form)
- Dolichocephaly (elongated head shape)
- Enlarged wrist and ankle joints
Advanced or Progressive Symptoms
Childhood Form (progression):
- Persistent or recurrent fractures
- Chronic bone pain
- Skeletal deformities worsening with growth
- Delayed puberty
- Dental complications (caries, periodontitis)
Adult Form (onset >18 years):
- Stress fractures, especially of metatarsals
- Thigh pain from femoral pseudofractures
- Chondrocalcinosis (calcium pyrophosphate deposition in joints)
- Osteoarthropathy
- Crystal arthropathies
- Enthesopathy (calcification of ligaments and tendons)
- Pseudogout
- Chronic musculoskeletal pain
- Premature loss of adult dentition
- Osteopenia or osteoporosis
Odontohypophosphatasia:
- Exclusively dental manifestations without skeletal involvement
- Premature exfoliation of primary and/or permanent teeth
- Severe dental caries
- Abnormal dentition
- Reduced alveolar bone
Common vs. Rare Symptoms
Common Symptoms (across multiple forms):
- Low alkaline phosphatase activity in serum
- Bone pain
- Dental abnormalities
- Fractures
- Delayed motor development (in pediatric forms)
Less Common Symptoms:
- Nephrocalcinosis (kidney calcification)
- Hypercalciuria
- Muscle weakness
- Growth failure
- Craniosynostosis
- Vitamin B6-responsive seizures
Rare Symptoms/Complications:
- Pseudotumor cerebri
- Peripheral neuropathy
- Hearing loss
- Ophthalmological complications (corneal calcification)
- Gastrointestinal problems
- Psychiatric manifestations (anxiety, depression)
- Cardiovascular calcifications
Symptom Progression Over Time
Perinatal Lethal Form:
- Rapidly progressive with death in utero or shortly after birth without treatment
- With treatment, possible survival with significant disability
Perinatal Benign Form:
- Initial severe presentation followed by spontaneous improvement
- May develop childhood or adult symptoms later in life
- Long-term outcome variable
Infantile Form:
- High mortality in first year of life without treatment (approximately 50%)
- With survival, progression to features of childhood form
- Permanent skeletal deformities common
- Neurodevelopmental sequelae in some survivors
Childhood Form:
- Symptoms often improve during puberty due to growth plate closure
- May remit temporarily and recur in adulthood
- Dental problems often persist
- Premature osteoarthritis in adulthood
Adult Form:
- Typically stable or slowly progressive
- May have been undiagnosed childhood form with temporary remission
- Increased fracture risk throughout life
- Progressive dental problems
- Joint complications often worsen with age
Odontohypophosphatasia:
- Usually stable with predominantly dental issues
- Some patients may develop mild skeletal symptoms later in life
Special Considerations for Symptom Expression
Several factors influence symptom expression in hypophosphatasia:
Genotype-Phenotype Correlation:
- Recessive mutations with two null alleles typically cause severe forms
- Dominant-negative mutations often associated with milder forms
- Compound heterozygosity can produce variable phenotypes
Environmental Factors:
- Physical activity levels may influence fracture risk
- Nutrition and vitamin D status may modify symptom severity
- Medications affecting bone metabolism may worsen symptoms
Age-related Changes:
- Growth spurts may worsen symptoms in children
- Puberty often brings temporary improvement
- Aging may unmask or exacerbate adult symptoms
Sex Differences:
- Pregnancy and lactation may unmask or worsen symptoms in women
- Possible hormonal influences on expression
The symptom profile of hypophosphatasia is notable for its extreme variability, even within families carrying the same mutation. This suggests the importance of genetic modifiers, environmental factors, and possibly epigenetic influences in determining disease severity and progression.
4. Causes
Genetic Basis
Hypophosphatasia is caused by mutations in the ALPL gene (also known as TNSALP), which is located on chromosome 1p36.1-p34 and spans approximately 50 kb containing 12 exons. This gene encodes the tissue-nonspecific alkaline phosphatase enzyme, which is expressed predominantly in the liver, bones, and kidneys.
Mutation Characteristics:
Mutation Diversity:
- More than 400 distinct mutations have been identified in the ALPL gene
- Most are missense mutations (approximately 75%)
- Other types include nonsense mutations, small deletions/insertions, splice site mutations, and regulatory region mutations
Mutation Distribution:
- Mutations occur throughout the gene with some clustering in exons 5, 6, 7, and 9
- Certain mutations show ethnic or geographic clustering
- Some mutations are recurrent in specific populations:
- c.1559delT in Japanese populations
- p.E174K (c.520G>A) in European populations
- c.571G>A in Manitoba Mennonites
Functional Consequences:
- Loss-of-function mutations result in reduced enzymatic activity
- Some mutations affect protein folding, intracellular transport, or stability
- Others affect substrate binding or catalytic activity
- Some mutations impair homodimer formation, essential for enzyme function
Inheritance Patterns
Hypophosphatasia follows two main inheritance patterns, with correlation to disease severity:
Autosomal Recessive Inheritance:
- Both ALPL alleles carry mutations
- Associated with severe forms (perinatal lethal, infantile, and many childhood cases)
- Parents are typically asymptomatic carriers with approximately 50% enzyme activity
- 25% risk of affected offspring when both parents are carriers
Autosomal Dominant Inheritance:
- One mutated ALPL allele causes disease
- Associated with milder forms (adult and odontohypophosphatasia)
- Often involves dominant-negative mutations where the mutant protein interferes with the function of the normal protein in the tetrameric enzyme
- 50% risk of transmission to offspring
Compound Heterozygosity:
- Two different mutations, one on each ALPL allele
- Phenotype depends on the combined effect of both mutations
- Common in patients with intermediate severity
Molecular Pathophysiology
The pathophysiology of hypophosphatasia involves several interrelated mechanisms:
Enzymatic Dysfunction:
- TNSALP normally hydrolyzes several substrates including:
- Inorganic pyrophosphate (PPi)
- Pyridoxal 5′-phosphate (PLP, vitamin B6)
- Phosphoethanolamine (PEA)
- TNSALP normally hydrolyzes several substrates including:
Mineralization Inhibition:
- Reduced TNSALP activity leads to extracellular accumulation of PPi
- PPi is a potent inhibitor of hydroxyapatite crystal formation
- Elevated PPi/Pi ratio directly impairs mineralization of bones and teeth
- This creates the central paradox of HPP: despite normal or elevated calcium and phosphate levels, mineralization is impaired
Vitamin B6 Metabolism Disruption:
- Elevated PLP levels outside cells
- Insufficient dephosphorylation of PLP to pyridoxal prevents vitamin B6 entry into cells
- Functional vitamin B6 deficiency in the brain may cause seizures in severe forms
Cellular and Tissue Effects:
- Abnormal osteoblast differentiation and function
- Disrupted chondrocyte maturation at growth plates
- Impaired collagen mineralization
- Potential osteocyte dysfunction
- Dysregulation of bone remodeling
Structural Consequences:
- Hypomineralized matrix in bones and teeth
- Rachitic changes at growth plates
- Osteomalacia in mature bone
- Weakened bone structure predisposing to fractures
- Dental abnormalities including defective cementum
Environmental and Modifying Factors
While hypophosphatasia is fundamentally a genetic disorder, several factors can modify its expression:
Dietary and Nutritional Factors:
- Excessive calcium intake may exacerbate hypercalcemia
- Vitamin D supplementation requires careful management (may worsen hypercalcemia)
- Zinc status may influence enzyme activity (TNSALP is a zinc-dependent enzyme)
Medications:
- Bisphosphonates are contraindicated as they are analogues of PPi and may worsen the condition
- Glucocorticoids can affect bone metabolism and potentially worsen bone quality
- Anti-epileptic drugs may interact with vitamin B6 metabolism
Physiological States:
- Pregnancy and lactation increase skeletal demands and may unmask or worsen symptoms
- Growth spurts may increase fracture risk in children
- Aging-related bone loss may compound HPP-related osteomalacia
Genetic Modifiers:
- Variations in other genes involved in mineralization may modify severity
- Polymorphisms in vitamin D receptor, collagen, and other bone-related genes
Activity Level and Mechanical Factors:
- High-impact activities may increase fracture risk
- Mechanical loading influences bone remodeling and symptom expression
Intercurrent Illnesses:
- Chronic inflammation may affect bone metabolism
- Immobilization can contribute to bone loss
Unlike many other genetic disorders, there are no known environmental triggers that precipitate the onset of hypophosphatasia. However, the factors listed above can significantly influence disease severity, progression, and complications, explaining some of the phenotypic variability observed even among patients with identical ALPL mutations.
5. Risk Factors
Genetic Risk Factors
The primary risk factors for hypophosphatasia are genetic, given its nature as an inherited disorder:
Family History:
- Having one or both parents who are carriers or affected
- Siblings with HPP
- Extended family history of HPP or unexplained bone disorders
- Family history of early tooth loss, unexplained fractures, or skeletal deformities
Consanguinity:
- Parental consanguinity (related parents) significantly increases risk for severe forms
- Particularly relevant in communities with high rates of consanguineous marriage
- Increases likelihood of inheriting two copies of the same recessive mutation
Ethnic Background:
- Certain populations have higher carrier frequencies:
- Manitoba Mennonites (1 in 25 carrier rate)
- Japanese populations (carrier frequency approximately 1 in 480)
- Higher prevalence in Caucasian populations compared to African or Asian populations
- Specific founder mutations in certain regional populations
- Certain populations have higher carrier frequencies:
Specific ALPL Mutations:
- Certain mutations confer higher risk for specific forms of HPP
- Null or severe mutations in both alleles correlate with perinatal or infantile forms
- Specific dominant-negative mutations associated with milder forms
- Some mutations have incomplete penetrance, resulting in variable expression
Age and Sex Considerations
Age-Related Risk:
- Different forms have characteristic age of onset:
- Perinatal: In utero to birth
- Infantile: Birth to 6 months
- Childhood: 6 months to 18 years
- Adult: 18+ years
- No age group is immune, as symptoms can present throughout the lifespan
- Some patients experience temporary symptom improvement during puberty with recurrence in adulthood
- Different forms have characteristic age of onset:
Sex-Related Factors:
- No clear sex predilection for inheritance (autosomal inheritance)
- Some evidence suggests females may be more likely to manifest symptoms in milder forms
- Pregnancy and lactation can unmask previously asymptomatic disease in women
- Sex hormones may modulate disease expression through effects on bone metabolism
Environmental and Lifestyle Modifiers
While not primary risk factors, these elements may modify disease expression in individuals with ALPL mutations:
Physical Activity:
- High-impact activities may increase fracture risk in affected individuals
- Extremely sedentary lifestyle may accelerate bone loss
- Appropriate exercise may help maintain bone strength and muscle support
Nutritional Factors:
- Extreme vitamin D deficiency or excess may influence symptom expression
- Calcium intake needs careful management
- Malnutrition may exacerbate bone fragility
Medications:
- Use of bisphosphonates can worsen HPP by further inhibiting mineralization
- Glucocorticoids may accelerate bone loss
- Anti-seizure medications may interact with vitamin B6 metabolism
Pre-existing Conditions and Risk Modification
Conditions That May Influence HPP Expression:
- Other genetic bone disorders (compound effect on bone strength)
- Endocrine disorders affecting bone metabolism:
- Thyroid dysfunction
- Growth hormone abnormalities
- Parathyroid disorders
- Chronic kidney disease (affects mineral metabolism)
- Chronic inflammatory conditions (affect bone remodeling)
- Vitamin D disorders
Pregnancy-Related Considerations:
- Pregnancy often exacerbates symptoms in women with HPP
- Increased skeletal calcium demands during pregnancy and lactation
- Hormonal changes affecting bone metabolism
- Higher risk of vitamin D deficiency during pregnancy
Risk Assessment and Genetic Counseling
Carrier Testing:
- Available for relatives of affected individuals
- Measurement of serum ALP activity (may be normal or slightly reduced in carriers)
- Genetic testing for known familial mutations
Recurrence Risk Assessment:
- For autosomal recessive forms:
- 25% risk when both parents are carriers
- Nearly 100% risk if both parents are affected (rare)
- 50% risk if one parent is affected (compound heterozygote) and the other is a carrier
- For autosomal dominant forms:
- 50% risk if one parent is affected
- For autosomal recessive forms:
Prenatal Risk Assessment:
- Prenatal diagnosis through:
- Chorionic villus sampling (10-12 weeks)
- Amniocentesis (15-20 weeks)
- Detailed ultrasound (can detect severe skeletal manifestations)
- Preimplantation genetic diagnosis possible for planned pregnancies through IVF
- Prenatal diagnosis through:
Risk Stratification:
- Genotype analysis can help predict severity in many cases
- Biochemical markers may supplement genetic information
- Family history patterns provide additional risk information
Understanding the risk factors for hypophosphatasia is essential for appropriate genetic counseling, family planning, early diagnosis, and management of the condition. The interplay between genetic background and environmental modifiers explains some of the phenotypic variability observed in this disorder and highlights the importance of personalized risk assessment and management approaches.
6. Complications
Skeletal Complications
Skeletal complications are the hallmark of hypophosphatasia and vary by disease form:
Fractures and Bone Fragility:
- Recurrent long bone fractures
- Insufficiency fractures with minimal trauma
- Pseudofractures (Looser’s zones), particularly in femurs
- Delayed or impaired fracture healing
- Chronic bone pain due to microfractures
Skeletal Deformities:
- Bowing of weight-bearing long bones
- Thoracic deformities (rachitic rosary, Harrison’s groove)
- Pectus excavatum or carinatum
- Scoliosis, kyphosis, and other spinal deformities
- Joint deformities and contractures
- Growth disturbances leading to short stature
Cranial Abnormalities:
- Craniosynostosis (premature fusion of skull sutures)
- Increased intracranial pressure
- Frontal bossing
- Dolichocephaly (elongated skull)
- Soft skull (caput membranaceum) in severe infantile forms
- Wormian bones (extra bones within cranial sutures)
Dental Complications
Dental problems are highly prevalent and often the presenting sign in milder forms:
Primary Dentition Issues:
- Premature loss of primary teeth (pathognomonic when teeth exfoliate with intact roots)
- Enamel hypoplasia
- Enlarged pulp chambers
- Abnormal dentin formation
Permanent Dentition Problems:
- Early loss of adult teeth
- Increased dental caries susceptibility
- Periodontal disease
- Root resorption
Supporting Structure Abnormalities:
- Reduced alveolar bone
- Cement hypoplasia
- Taurodontism (enlarged pulp chambers)
- Malocclusion due to premature tooth loss
Neurological Complications
Neurological complications are most common in severe perinatal and infantile forms:
Seizures:
- Vitamin B6-responsive seizures due to impaired cellular uptake of vitamin B6
- Occur in approximately 20-30% of infants with severe disease
- Can be refractory to standard anticonvulsants
Increased Intracranial Pressure:
- Secondary to craniosynostosis
- Can lead to developmental delays if untreated
- May require neurosurgical intervention
Other Neurological Issues:
- Developmental delays
- Motor skill impairment
- Headaches
- Rarely, spinal cord compression from vertebral fractures
- Chiari malformation (reported in some cases)
Respiratory Complications
Respiratory problems are life-threatening in severe forms:
Respiratory Insufficiency:
- Primary cause of mortality in perinatal and infantile forms
- Due to poorly mineralized rib cage and rachitic deformities
- Pulmonary hypoplasia in severe perinatal cases
- Restrictive lung disease from chest deformities
Pneumonia and Respiratory Infections:
- Secondary to inadequate chest expansion
- Compounded by difficulty clearing secretions
- Leading cause of death in untreated infantile form
Sleep-Disordered Breathing:
- Obstructive and central sleep apnea
- Contributes to daytime fatigue and cognitive issues
Metabolic and Systemic Complications
Mineral Metabolism Disturbances:
- Hypercalcemia (elevated serum calcium)
- Hypercalciuria (excessive urinary calcium excretion)
- Nephrocalcinosis (kidney calcification)
- Renal function impairment secondary to calcifications
Muscular Complications:
- Generalized hypotonia, particularly in infants
- Muscle weakness
- Delayed motor milestone achievement
- Gait abnormalities
Growth and Development Issues:
- Failure to thrive in infantile forms
- Growth retardation and short stature
- Delayed puberty in some cases
- Impaired physical capabilities
Psychosocial Complications
The chronic nature of HPP often leads to significant psychosocial impacts:
Psychological Issues:
- Chronic pain leading to depression and anxiety
- Body image concerns related to skeletal deformities
- Adjustment disorders
- Developmental and cognitive challenges in severe childhood-onset cases
Social and Quality of Life Impacts:
- Educational disruptions due to hospitalizations
- Employment limitations in adulthood
- Reduced participation in physical activities
- Financial burden from medical expenses
- Caregiver stress in families with affected children
Long-term Disability and Mortality
The potential for disability and mortality varies significantly by disease form:
Perinatal Lethal Form:
- Nearly 100% mortality without treatment
- With treatment, survival possible but with significant disability
- Respiratory failure is the primary cause of death
Infantile Form:
- Historical mortality rate of approximately 50% within the first year
- Dramatically improved with enzyme replacement therapy
- Survivors often have permanent skeletal deformities and developmental challenges
Childhood Form:
- Rarely life-threatening
- Can lead to significant disability from fractures, pain, and growth disturbance
- Often improves during puberty but may recur in adulthood
Adult Form:
- Normal life expectancy in most cases
- Chronic pain and mobility issues common
- Fracture-related complications can cause significant morbidity
- Quality of life often affected by pain and functional limitations
Odontohypophosphatasia:
- No effect on life expectancy
- Significant dental morbidity
- Psychological impact of dental health issues
Disease Burden and Healthcare Impact
The complications of hypophosphatasia create substantial healthcare utilization:
Hospitalization Patterns:
- Multiple hospitalizations for fracture management
- Respiratory support in severe forms
- Surgical interventions for skeletal deformities and craniosynostosis
Multidisciplinary Care Requirements:
- Ongoing need for coordinated care across specialties
- Regular monitoring for complications
- Rehabilitative services
Impact of New Therapies:
- Enzyme replacement therapy has dramatically reduced mortality in severe forms
- Shifted the paradigm from palliative to therapeutic approach
- Transformed previously fatal forms into chronic conditions requiring ongoing management
The complications of hypophosphatasia create a complex clinical picture requiring vigilant monitoring and proactive management. The availability of enzyme replacement therapy has significantly altered the natural history of severe forms, but patients across the spectrum still face substantial morbidity and need comprehensive, multidisciplinary care to optimize outcomes and quality of life.
7. Diagnosis & Testing
Clinical Diagnostic Approach
The diagnosis of hypophosphatasia requires a combination of clinical awareness, biochemical testing, radiographic evaluation, and genetic confirmation. The diagnostic approach typically follows this pathway:
Clinical Suspicion based on:
- Skeletal manifestations (fractures, deformities)
- Dental issues (premature tooth loss with intact roots)
- Family history of bone disease
- Poor response to conventional treatment for rickets or osteomalacia
- Presence of characteristic features based on age of presentation
Differential Diagnosis consideration:
- Other forms of rickets and osteomalacia
- Osteogenesis imperfecta
- Cleidocranial dysplasia
- Hypophosphatemic rickets
- Primary hypoparathyroidism
- Nutritional rickets
Diagnostic Algorithm:
- Initial serum ALP measurement
- If low, proceed to substrate testing and genetic analysis
- Radiographic evaluation
- Exclusion of other conditions with similar presentations
Biochemical Testing
Key Laboratory Tests:
Alkaline Phosphatase (ALP) Activity:
- Cornerstone of diagnosis
- Must be interpreted using age and sex-specific reference ranges
- Typically markedly reduced (often <50% of lower limit of normal)
- May be only moderately reduced in milder forms
- Important: Values should be assessed before treatment with bisphosphonates or other bone-active agents, which can lower ALP
TNSALP Substrates:
- Elevated substrates provide supporting evidence:
- Plasma pyridoxal 5′-phosphate (PLP) – most sensitive and specific
- Urinary phosphoethanolamine (PEA)
- Plasma or urinary inorganic pyrophosphate (PPi) – technically challenging to measure
- Elevated substrates provide supporting evidence:
Other Relevant Tests:
- Serum calcium (may be elevated, especially in infants)
- Urine calcium (hypercalciuria common)
- Serum phosphate (usually normal or slightly elevated)
- Parathyroid hormone (typically normal or slightly suppressed if hypercalcemic)
- Vitamin D metabolites (to exclude vitamin D deficiency)
Genetic Testing
Genetic analysis is essential for confirming diagnosis, determining inheritance pattern, and assessing recurrence risk:
Molecular Genetic Testing Options:
- Targeted mutational analysis for common mutations
- Single-gene sequencing of ALPL
- Gene panel testing for skeletal dysplasias
- Next-generation sequencing approaches
- Deletion/duplication analysis if point mutations aren’t identified
Genetic Testing Indications:
- Confirmation of biochemical diagnosis
- Prenatal diagnosis
- Carrier testing in families
- Cases with borderline biochemical results
- Distinguishing dominant vs. recessive forms
Genotype-Phenotype Correlation:
- Helps predict disease severity and progression
- Informs genetic counseling
- Certain mutations correlate with specific clinical forms
- Compound heterozygotes often have intermediate phenotypes
Radiographic Evaluation
Imaging studies are crucial for diagnosis and monitoring:
Plain Radiographs:
- Show characteristic features depending on age:
- In severe forms: Profound hypomineralization, shortened limbs, ossification defects
- In infantile form: Rachitic changes at metaphyses, irregular provisional zones of calcification, “tongues” of radiolucency extending into metaphyses
- In childhood/adult forms: Osteopenia, stress fractures, pseudofractures
- Show characteristic features depending on age:
Specific Radiographic Findings:
- Cranial: Wormian bones, delayed suture closure, or craniosynostosis
- Spine: Osteopenia, fractures, scoliosis
- Long bones: Bowing, fractures, widened metaphyses
- Ribs: Fractures, rachitic rosary appearance
- Femur: Characteristic pseudofractures (Looser’s zones) on lateral aspect
Advanced Imaging:
- Bone densitometry (DXA) – typically shows reduced bone mineral density
- CT – useful for evaluating craniosynostosis and complex fractures
- MRI – helps assess growth plates and complications like spinal cord compression
Prenatal and Neonatal Diagnosis
Early diagnosis is critical, particularly for severe forms:
Prenatal Diagnostic Methods:
- Chorionic villus sampling (10-12 weeks) for genetic analysis
- Amniocentesis (15-20 weeks) for genetic analysis
- Detailed ultrasound may detect skeletal abnormalities in severe forms
- 3D ultrasound enhances detection of subtle skeletal changes
- Maternal serum ALP is not helpful (placental ALP predominates)
Preimplantation Genetic Diagnosis:
- Option for families with known mutations
- Performed in conjunction with in vitro fertilization
- Allows selection of unaffected embryos
Newborn Screening:
- Not routinely performed for HPP
- Low ALP activities may be incidentally noted
- Caution needed as premature infants can have transiently low ALP
Special Diagnostic Considerations
Mild Form Detection Challenges:
- May be overlooked due to subtle presentation
- Consider in adults with recurrent stress fractures, especially metatarsal
- Dental professionals should be aware of dental manifestations
- Low ALP may be incidentally discovered on routine testing
Age-Specific Reference Ranges:
- Critical for ALP interpretation
- ALP naturally higher in infants, children, and during puberty
- Pregnancy and other conditions can elevate ALP
- False negatives may occur if inappropriate reference ranges are used
Diagnostic Pitfalls:
- Misdiagnosis as osteogenesis imperfecta or other bone disorders
- Delayed diagnosis of milder forms
- Failure to recognize HPP when dental manifestations are the primary presentation
- Misinterpretation of ALP levels due to incorrect reference ranges
- Non-specific symptoms (pain, fatigue) in adult forms leading to delayed diagnosis
Diagnostic Approaches by Life Stage:
- Prenatal: Genetic testing and specialized ultrasound
- Infancy: Biochemical testing, radiographs, and genetic confirmation
- Childhood: Focus on dental manifestations and fracture patterns
- Adulthood: Consider in context of low-trauma fractures and persistent pain
The diagnosis of hypophosphatasia requires a high index of suspicion, careful biochemical testing with appropriate reference ranges, radiographic evaluation, and genetic confirmation. Early and accurate diagnosis is essential for appropriate management, particularly with the availability of enzyme replacement therapy for severe forms. The wide spectrum of disease severity makes diagnosis challenging in milder cases, necessitating awareness of subtle manifestations and appropriate use of diagnostic tools.
8. Treatment Options
Enzyme Replacement Therapy
Enzyme replacement therapy (ERT) has revolutionized the treatment of severe hypophosphatasia:
Asfotase Alfa (Strensiq®):
- First disease-specific treatment for HPP
- Recombinant human TNSALP enzyme with bone-targeting domain
- FDA and EMA approved in 2015
- Indications: Perinatal/infantile and juvenile-onset HPP
- Dosing: 2 mg/kg three times weekly or 1 mg/kg six times weekly, subcutaneous injection
- Mechanism: Replaces deficient TNSALP enzyme activity, enabling proper mineralization
Efficacy in Different Patient Groups:
Perinatal/Infantile Form:
- Dramatic improvement in survival (survival rate >95% vs. historical <50%)
- Improved respiratory function
- Enhanced bone mineralization
- Reduced need for respiratory support
- Improved growth parameters
Childhood Form:
- Improved height and weight gain
- Reduced fracture rates
- Improved mobility
- Reduced pain
- Radiographic evidence of healing of rickets
Adult Form:
- Not FDA-approved but under investigation
- Case reports suggest benefit for fracture healing
- Potential improvement in pain and function
Monitoring Treatment Response:
- Regular radiographic assessment
- Growth parameters in children
- Functional mobility assessments
- Respiratory function in severe cases
- Quality of life measures
- Biochemical markers (though ALP levels will be elevated due to the treatment)
Treatment Challenges:
- High cost ($200,000-$1,500,000 per year depending on weight)
- Need for frequent injections
- Lifelong therapy requirement
- Injection site reactions (most common adverse effect)
- Hypersensitivity reactions (including anaphylaxis)
- Limited data on long-term outcomes
- Optimal treatment duration still being determined
Supportive and Symptomatic Treatments
Before ERT and as adjunctive therapy, several supportive approaches are important:
Pain Management:
- Non-opioid analgesics (acetaminophen, NSAIDs with caution)
- Physical therapy modalities
- Heat and cold therapy
- In severe cases, opioid analgesics for breakthrough pain
- Evaluation for specific sources (fractures, pseudofractures)
Fracture Management:
- Careful orthopedic care
- Sometimes non-standard approaches required due to poor healing
- Avoidance of prolonged immobilization
- Consideration of non-operative management when possible
- Special considerations for hardware placement in soft bone
Calcium and Vitamin D Management:
- Careful monitoring of calcium levels
- Avoidance of supplementation if hypercalcemic
- Potential need for restriction in hypercalcemic infants
- Judicious vitamin D supplementation
Dental Care:
- Early and regular dental evaluations
- Preventive dental care to preserve teeth
- Prompt management of loose teeth
- Prosthetic replacement of lost teeth
- Special attention to oral hygiene
- Consideration of dental implants (requires evaluation of bone quality)
Respiratory Support:
- Supplemental oxygen
- Ventilatory support as needed
- Pulmonary hygiene and secretion clearance
- Prevention and aggressive treatment of respiratory infections
- Pulmonary rehabilitation
Surgical Interventions
Various surgical procedures may be necessary to address specific complications:
Neurosurgical Procedures:
- Cranial vault expansion for craniosynostosis
- Ventricular shunting for hydrocephalus
- Management of Chiari malformation if present
Orthopedic Surgeries:
- Correction of significant deformities
- Rodding procedures for recurrent fractures
- Osteotomies to realign bones
- Management of non-unions
- Special considerations for bone quality and healing potential
Dental and Maxillofacial Procedures:
- Specialized dental restorations
- Considerations for orthodontic treatment
- Prosthodontic rehabilitation
- Dental implants in appropriate cases
Therapeutic Approaches by Disease Form
Different clinical forms require tailored approaches:
Perinatal Lethal Form:
- Immediate initiation of asfotase alfa if diagnosed prenatally or at birth
- Intensive respiratory support
- Careful nutritional management
- Multidisciplinary team approach including neonatology, genetics, orthopedics, and pulmonology
Infantile Form:
- Early initiation of asfotase alfa
- Management of hypercalcemia if present
- Vitamin B6 for seizure control
- Careful monitoring of growth and development
- Nutritional support
- Early intervention services
Childhood Form:
- Evaluation for asfotase alfa eligibility
- School accommodations for physical limitations
- Dental monitoring and management
- Physical therapy for strength and function
- Pain management strategies
- Psychosocial support
Adult Form:
- Focus on pain management
- Fracture prevention
- Physical therapy and exercise guidance
- Dental care
- Consideration of off-label asfotase alfa in severe cases
Odontohypophosphatasia:
- Primarily dental management
- Regular dental monitoring
- Early intervention for dental issues
- Prosthodontic rehabilitation as needed
Rehabilitative and Supportive Interventions
Comprehensive care includes various non-pharmacological approaches:
Physical Therapy:
- Strengthening exercises
- Gait training
- Assistance with mobility aids
- Balance training
- Aquatic therapy (reduced weight-bearing stress)
- Guidance on appropriate physical activity levels
Occupational Therapy:
- Activities of daily living assessment and training
- Adaptive equipment recommendations
- Energy conservation techniques
- Home and school/workplace modifications
Nutritional Management:
- Balanced diet with appropriate calcium
- Monitoring for vitamin D status
- Weight management to reduce skeletal stress
- Adequate protein for bone health
- Avoidance of excessive phosphate (especially in supplements)
Psychological Support:
- Counseling for adjustment to chronic condition
- Pain coping strategies
- Support groups
- Assessment for depression and anxiety
- School and career counseling
Emerging and Investigational Therapies
Several promising approaches are in development:
Gene Therapy:
- Delivery of functional ALPL gene
- Viral vector approaches in preclinical development
- Potential for long-term correction of enzyme deficiency
Bone-Targeted Enzyme Replacement:
- New formulations with improved half-life
- Alternative delivery methods
- More efficient bone-targeting strategies
Small Molecule Approaches:
- Compounds to enhance residual enzyme activity
- Chaperone therapies to improve protein folding
- Anti-sclerostin antibodies to promote bone formation
Alternative Management Strategies:
- RANKL inhibitors to reduce bone turnover
- Calcium-sensing receptor modulators for hypercalcemia
- Novel pain management approaches
Cell-Based Therapies:
- Mesenchymal stem cell transplantation
- Gene-modified cell therapies
- Bone marrow transplantation (limited success to date)
Multidisciplinary Care Approach
Optimal management requires coordination across specialties:
Core Team Members:
- Metabolic bone specialist/endocrinologist
- Medical geneticist
- Orthopedic surgeon
- Dentist/dental specialist
- Physical therapist
- Genetic counselor
Additional Specialists as Needed:
- Neurosurgeon (for craniosynostosis)
- Pulmonologist (for respiratory complications)
- Nephrologist (for renal complications)
- Pain specialist
- Rheumatologist
- Neurologist (for seizure management)
Care Coordination:
- Regular multidisciplinary clinics when possible
- Shared care protocols
- Transition planning from pediatric to adult care
- Patient registry participation
The management of hypophosphatasia has been transformed by the introduction of enzyme replacement therapy, particularly for severe forms. However, comprehensive care still requires a multidisciplinary approach addressing all aspects of this complex disorder. Treatment must be individualized based on disease severity, complications, and patient-specific factors, with ongoing monitoring and adjustment throughout the lifespan.
9. Prevention & Precautionary Measures
Genetic Counseling and Family Planning
As a genetic disorder, primary prevention of hypophosphatasia centers on genetic counseling and reproductive options:
Genetic Counseling Process:
- Risk assessment based on family history
- Discussion of inheritance patterns
- Explanation of reproductive options
- Psychological support for decision-making
- Information about disease severity spectrum
Carrier Testing:
- Molecular genetic testing for known familial mutations
- Serum ALP activity (may be normal or slightly reduced in heterozygotes)
- Offered to relatives of affected individuals
- Particularly important for siblings of affected children and extended family in autosomal recessive forms
Reproductive Options for At-Risk Couples:
- Prenatal Diagnosis:
- Chorionic villus sampling (10-12 weeks)
- Amniocentesis (15-20 weeks)
- Early detailed ultrasound for severe forms
- Preimplantation Genetic Diagnosis (PGD):
- Testing embryos created through in vitro fertilization
- Selection of unaffected embryos for transfer
- Option for couples at high risk of having an affected child
- Gamete Donation:
- Use of donor eggs or sperm depending on which partner carries mutation(s)
- Eliminates risk if donor is screened
- Adoption:
- Alternative family-building option
- Prenatal Diagnosis:
Cascade Testing in Families:
- Systematic approach to identify at-risk relatives
- Testing moving outward from the proband (affected individual)
- Particularly important for extended family in recessive forms
- May identify mildly affected individuals who were previously undiagnosed
Precautionary Measures for Diagnosed Individuals
While hypophosphatasia cannot be prevented once a person is affected, several precautions can minimize complications:
Medication Precautions:
- Absolute Contraindications:
- Bisphosphonates (may worsen mineralization defects)
- High-dose fluoride treatments
- Use with Caution:
- Glucocorticoids (may worsen bone quality)
- NSAIDs (may affect renal function if already compromised)
- Some anticonvulsants (interaction with vitamin B6 metabolism)
- Medication Alerts:
- Medical alert bracelet
- Clear documentation in medical records
- Patient education about medications to avoid
- Absolute Contraindications:
Dental Precautions and Preventive Care:
- Early and regular dental evaluations (every 3-6 months)
- Meticulous oral hygiene
- Preventive sealants and fluoride treatments
- Prompt attention to loose teeth
- Careful planning of dental procedures
- Consideration of prophylactic antibiotics for dental work (if history of endocarditis risk)
Fracture Prevention Strategies:
- Appropriate physical activity guidance
- Fall prevention measures
- Home safety assessment
- Proper footwear
- Assistive devices when necessary
- Avoidance of high-impact activities in more severe forms
Nutritional Guidance:
- Balanced calcium intake (avoiding excess)
- Appropriate vitamin D levels
- Adequate but not excessive phosphate
- Protein sufficiency for bone health
- Avoidance of extreme diets
- Weight management to reduce skeletal stress
Monitoring and Surveillance
Regular monitoring can identify complications early and guide intervention:
Clinical Monitoring Schedule:
- Severe forms: Monthly to quarterly visits initially
- Childhood form: Every 3-6 months
- Adult form: Every 6-12 months
- More frequent during growth periods, pregnancy, or with complications
Growth and Development Monitoring:
- Regular height and weight measurements
- Developmental assessments in children
- Tracking of motor milestones
- School performance evaluation
Imaging Surveillance:
- Skeletal surveys at diagnosis
- Follow-up radiographs based on clinical needs
- Bone density testing (DXA) every 1-2 years
- Renal ultrasound if history of hypercalciuria or nephrocalcinosis
Laboratory Monitoring:
- Calcium levels (serum and urine)
- Renal function tests
- Vitamin D status
- ALP substrates to monitor disease activity
- In patients on ERT, monitoring for antibody development
Special Considerations for Life Stages
Different life stages require specific preventive approaches:
Infancy and Childhood:
- Early intervention services
- Physical therapy to optimize motor development
- Educational accommodations
- Regular dental monitoring
- Growth monitoring
- Nutritional guidance
- Immunizations per standard schedules
Adolescence:
- Guidance during growth spurts
- Dental and orthodontic planning
- Career and education counseling considering physical limitations
- Transition planning to adult care
- Education about inheritance
Adulthood:
- Occupational adaptations
- Ergonomic assessments
- Pregnancy planning and management
- Ongoing dental care
- Regular bone health monitoring
- Pain management strategies
Pregnancy:
- Pre-conception counseling
- Increased monitoring during pregnancy
- Calcium management
- Consideration of delivery method
- Special dental attention
- Post-partum monitoring (increased risk of symptoms)
Environmental and Lifestyle Modifications
Environmental factors and lifestyle choices can modify disease expression:
Physical Activity Recommendations:
- Individualized exercise prescriptions
- Low-impact activities (swimming, cycling)
- Strengthening of muscles around susceptible joints
- Balance between activity and rest
- Avoidance of contact sports in more severe forms
Home and School/Workplace Adaptations:
- Ergonomic assessments
- Adaptive equipment
- Environmental modifications to prevent falls
- Consideration of reduced hours or modified duties if pain or fatigue significant
Stress Management and Mental Health:
- Psychological support
- Stress reduction techniques
- Pain management strategies
- Support groups
- Regular assessment for depression and anxiety
Educational Measures and Resources
Education of patients, families, and healthcare providers is essential:
Patient and Family Education:
- Disease information
- Inheritance patterns
- Recognition of complications
- Medication precautions
- When to seek medical attention
- Self-advocacy skills
Healthcare Provider Education:
- Primary care physicians
- Dentists
- Emergency department staff
- School nurses
- Creation of emergency protocols
Community Resources:
- Patient advocacy groups:
- Soft Bones, Inc. (US)
- The Hypophosphatasia Foundation (US)
- HPP Contact (UK)
- International patient organizations
- Financial assistance programs for medication costs
- Disability services information
- Educational advocacy resources
- Patient advocacy groups:
While hypophosphatasia cannot be prevented in those already affected, a comprehensive approach to precautionary measures, monitoring, and education can significantly reduce complications and improve quality of life. Genetic counseling and reproductive planning options provide important preventive strategies for future generations. The focus should be on individualized approaches based on disease severity, age, and specific needs, with regular reassessment and adaptation as the disease evolves or life circumstances change.
10. Global & Regional Statistics
Global Prevalence and Incidence
Hypophosphatasia is classified as a rare disease, but its prevalence varies significantly by population and disease form:
Overall Estimated Prevalence:
- Severe forms (perinatal and infantile): Approximately 1 in 100,000 live births globally
- Moderate forms (childhood): Approximately 1 in 6,370 to 1 in 20,000
- Mild forms (adult and odontohypophosphatasia): Likely underdiagnosed, estimated 1 in 6,000 to 1 in 900
- All forms combined: Estimated 1 in 2,500 to 1 in 10,000 depending on the population
Incidence Patterns:
- Higher detection rates in countries with advanced healthcare systems
- Increased recognition with the advent of genetic testing
- Higher carrier frequencies in certain populations due to founder effects
- Milder forms often diagnosed incidentally or retrospectively after diagnosis of a more severely affected family member
Challenges in Estimating Prevalence:
- Underdiagnosis of milder forms
- Variable clinical presentation leading to misdiagnosis
- Limited awareness among healthcare providers
- Lack of population-based studies in many regions
- Insufficient registry data in many countries
Regional Variations
Significant geographic and ethnic variations exist in HPP prevalence:
North America:
- United States: Estimated prevalence of severe forms 1 in 100,000
- Canada (Manitoba Mennonite population): 1 in 2,500 due to founder effect
- Canada (general population): Similar to US rates
Europe:
- United Kingdom: Estimated 1 in 6,370 based on carrier frequency studies
- France: Approximately 1 in 100,000 for severe forms
- Netherlands: 1 in 11,500 based on diagnostic registry data
- Scandinavia: Limited data, but appearance of founder mutations in certain regions
Asia:
- Japan: Higher reported prevalence of severe forms (1 in 60,000)
- Japan: Common founder mutation c.1559delT with carrier frequency of 1 in 480
- China: Limited population data, but case series suggest lower prevalence than in Caucasian populations
- South Korea: Few published cases, suggesting potentially lower prevalence
Middle East and North Africa:
- Higher rates in populations with consanguineous marriages
- Limited published epidemiological data
- Case series from Turkey suggest higher prevalence than global average
Latin America:
- Limited epidemiological studies
- Case series from Brazil and Argentina suggest prevalence similar to European populations
- Likely underdiagnosis in regions with limited healthcare access
Australia and New Zealand:
- Prevalence similar to European estimates
- Better recognition due to advanced healthcare systems
- Small population size limits statistical significance
Carrier Frequencies
Carrier frequencies have been studied in several populations:
- Manitoba Mennonites: 1 in 25 (highest known carrier rate)
- Japanese: 1 in 480 for the c.1559delT mutation
- European descent: 1 in 40 to 1 in 80
- African populations: Limited data, appears less common
- Asian populations outside Japan: Lower carrier frequencies than in European populations
Mortality and Survival Rates
Mortality varies dramatically by disease form and access to treatment:
Perinatal Lethal Form:
- Pre-treatment era: Nearly 100% mortality in utero or within weeks of birth
- With enzyme replacement therapy: Survival rates approaching 95% in treated cohorts
Infantile Form:
- Historical mortality: Approximately 50% within the first year of life
- Primary cause of death: Respiratory failure
- With modern care including ERT: >90% survival rate
- Long-term outcomes still being studied
Childhood Form:
- Rarely life-threatening
- Normal life expectancy with appropriate management
- Morbidity rather than mortality is the primary concern
Adult Form:
- Normal life expectancy
- Quality of life impact from fractures and pain
- Limited data on long-term outcomes
Impact of Enzyme Replacement Therapy:
- Transformative effect on survival in severe forms
- Long-term data still accumulating
- Access inequities impact survival rates globally
Disease Burden and Healthcare Economics
The economic and healthcare burden of HPP varies by region and severity:
Direct Medical Costs:
- Enzyme replacement therapy: $200,000 to $1,500,000 per year depending on weight
- Hospitalization costs: Significant for severe forms
- Surgical interventions: Often multiple procedures throughout life
- Outpatient management: Multiple specialist visits
Indirect Costs:
- Caregiver burden
- Lost productivity
- Special education needs
- Home modifications
- Assistive devices
Regional Economic Disparities:
- Limited access to enzyme replacement therapy in many countries
- Disproportionate burden in regions without universal healthcare
- Orphan drug policies affecting accessibility
- Disparities in multidisciplinary care availability
Diagnostic Rates and Temporal Trends
Patterns of diagnosis have evolved over time:
Historical Trends:
- Pre-1980s: Only severe forms typically recognized
- 1980s-1990s: Increased recognition with biochemical testing
- 2000s-present: Expanded diagnosis with genetic testing
- Post-2015: Increased screening due to treatment availability
Current Diagnostic Patterns:
- Growing recognition of milder forms
- Earlier diagnosis of severe forms
- Incidental diagnoses through routine lab testing
- Retrospective diagnoses in families after index case identified
Diagnostic Delays:
- Average delay from symptom onset to diagnosis:
- Severe forms: Weeks to months
- Childhood form: 1-5 years
- Adult form: Often >10 years
- Multiple healthcare provider visits before diagnosis common in milder forms
- Average delay from symptom onset to diagnosis:
Registry Data and Research Patterns
Several registries are collecting data to better understand the global picture:
Global HPP Registry:
- International, industry-sponsored registry
- Collects natural history and treatment outcome data
- Currently includes patients from over 20 countries
National Registries:
- Japanese HPP Registry
- European registries (France, Germany, UK)
- US-based registries through patient organizations
Research Publication Trends:
- Increased publication rate since 2015 (post-ERT approval)
- Geographic disparities in research output
- Shift from descriptive to treatment-focused studies
- Growing focus on quality of life outcomes
Public Health Impact and Awareness
The recognition of HPP as a public health concern varies globally:
Orphan Disease Designation:
- Official orphan disease status in US, EU, and Japan
- Varying levels of supportive policies
- Impact on research funding and drug development
Screening Programs:
- No routine newborn screening for HPP
- Consideration of selective screening in high-risk populations
- Potential for future inclusion in expanded newborn screening
Awareness Initiatives:
- HPP Awareness Day (October 30)
- Patient advocacy organization activities
- Professional education initiatives
- Social media campaigns
The global landscape of hypophosphatasia demonstrates significant regional variations in prevalence, diagnosis, and management. While substantial progress has been made in understanding the epidemiology of HPP, many knowledge gaps remain, particularly regarding milder forms and prevalence in underrepresented populations. The advent of effective treatment has changed the natural history of severe forms but also highlighted disparities in access to diagnosis and care worldwide.
11. Recent Research & Future Prospects
Recent Advances in Understanding
Significant progress has been made in understanding hypophosphatasia at molecular, cellular, and clinical levels:
Molecular Insights:
- Expanded mutation database (>400 ALPL mutations cataloged)
- Improved understanding of dominant-negative effects in heterozygous mutations
- Recognition of the impact of specific mutations on enzyme stability, substrate binding, and protein trafficking
- Identification of genotype-phenotype correlations for certain mutations
Pathophysiological Mechanisms:
- Deeper understanding of tissue-nonspecific alkaline phosphatase’s role in:
- Hydroxyapatite crystal nucleation and growth
- Regulation of pyrophosphate/phosphate ratio
- Cross-talk with other mineralization pathways
- Recognition of TNSALP’s role beyond bone:
- Neural development and function
- Vitamin B6 metabolism
- Potential roles in immunity and inflammation
- Deeper understanding of tissue-nonspecific alkaline phosphatase’s role in:
Clinical Natural History:
- Better delineation of the disease spectrum
- Long-term follow-up data from registry studies
- Documented improvement in survival with treatment
- Recognition of previously unappreciated manifestations
Biomarker Development:
- Validation of PLP as a sensitive diagnostic and monitoring marker
- Exploration of novel biomarkers including:
- Osteoblast-derived matrix proteins
- Metabolomic profiles
- micro-RNAs associated with bone mineralization
Current Treatment Developments
Research on treatment continues to evolve beyond the initial approval of asfotase alfa:
Enzyme Replacement Therapy Optimization:
- Long-term safety and efficacy data collection
- Dosing optimization studies
- Extended dosing interval investigations
- Alternative administration routes exploration
- Development of improved bone-targeting strategies
Novel Formulations:
- Extended half-life versions
- Oral delivery systems in preclinical development
- Encapsulated enzyme formulations
- Nanotechnology-based delivery systems
Expanded Clinical Applications:
- Use in adult-onset HPP (currently off-label)
- Treatment during pregnancy
- Age-specific dosing protocols
- Treatment of specific complications (e.g., craniosynostosis)
Combinatorial Approaches:
- ERT combined with anti-sclerostin antibodies
- ERT with calcium-sensing receptor modulators
- Multimodal approaches for specific manifestations
Gene Therapy and Genetic Approaches
Perhaps the most exciting frontier in HPP research is gene therapy:
Viral Vector Approaches:
- Adeno-associated virus (AAV) delivery of functional ALPL gene
- Lentiviral vectors for ex vivo cell modification
- Bone marrow-directed gene therapy
- Liver-directed gene therapy with bone targeting of expressed enzyme
- Preclinical proof-of-concept demonstrated in mouse models
Gene Editing Technologies:
- CRISPR/Cas9 approaches for correction of specific mutations
- Base editing for point mutations
- Prime editing technology for precise gene correction
- Challenges related to delivery to bone tissue being addressed
mRNA Therapeutics:
- Delivery of ALPL mRNA for transient expression
- Potential for repeated administration
- Lipid nanoparticle delivery systems
- Bone-targeting modifications
Stem Cell-Based Approaches:
- Mesenchymal stem cell transplantation
- Gene-corrected autologous stem cells
- Induced pluripotent stem cells (iPSCs) as disease models and potential therapy
- Combined cell therapy and gene therapy approaches
Small Molecule Development
Several small molecule approaches are under investigation:
Pharmacological Chaperones:
- Small molecules that stabilize mutant TNSALP
- Potentially most effective for missense mutations affecting protein folding
- Several candidates in preclinical development
- May allow restoration of residual enzyme activity
Substrate Reduction Therapy:
- Compounds targeting pyrophosphate production
- Modulation of mineralization inhibitors
- Alternative approaches to manage substrate accumulation
Non-Catalytic Approaches:
- Calcium-sensing receptor modulators for hypercalcemia
- Anti-sclerostin antibodies to promote bone formation
- RANK-L inhibitors to reduce bone turnover
- Anabolic agents for severe osteoporosis in adult HPP
Repurposed Medications:
- Screening of approved drugs for ALPL-enhancing effects
- Exploration of drugs affecting related mineralization pathways
- Potential faster path to clinical approval
Clinical Research Initiatives
Multiple clinical research efforts are advancing understanding and treatment:
Natural History Studies:
- Global HPP Registry (industry-sponsored)
- National and regional registries
- Focus on milder forms previously understudied
- Long-term outcomes assessment
Clinical Trial Developments:
- Age-specific intervention studies
- Trials for adults with HPP
- Comparative effectiveness research
- Quality of life outcome measures
Diagnostic Advancements:
- Development of diagnostic algorithms
- Artificial intelligence applications for radiographic diagnosis
- Point-of-care testing for ALP and substrates
- Next-generation sequencing panels including ALPL
Real-world Evidence Collection:
- Post-marketing surveillance of asfotase alfa
- Observational studies of treated and untreated patients
- Patient-reported outcome measures
- Cost-effectiveness analyses
Future Prospects and Timeline
The future of HPP research and treatment holds significant promise:
Near-Term Prospects (1-5 years):
- Expanded indications for asfotase alfa
- Improved dosing protocols
- Better understanding of long-term outcomes
- Enhanced diagnostic algorithms
- First human trials of novel treatment approaches
Medium-Term Prospects (5-10 years):
- Alternative ERT formulations
- Pharmacological chaperones in clinical use
- Potential early gene therapy trials
- Biomarker-guided personalized treatment approaches
- Expanded newborn screening in high-risk populations
Long-Term Prospects (10+ years):
- Potentially curative gene therapy
- Combination approaches targeting multiple aspects of disease
- Prevention strategies for high-risk families
- Integration with broader bone health management
- Affordable treatment options for global access
Challenges and Barriers
Several challenges remain in advancing HPP research and treatment:
Scientific Challenges:
- Tissue-specific delivery of gene therapy
- Long-term safety of genetic approaches
- Development of treatment for neurological manifestations
- Understanding modifiers of disease expression
Clinical Implementation Barriers:
- Extreme cost of current ERT
- Limited access in many countries
- Diagnostic delays and misdiagnosis
- Transition from pediatric to adult care
Research Infrastructure Needs:
- More comprehensive registries
- Biobanks for sample collection
- International collaboration
- Patient engagement in research prioritization
Knowledge Gaps:
- Natural history of milder forms
- Optimal management of adults
- Treatment during pregnancy
- Non-skeletal manifestations
The research landscape for hypophosphatasia has been transformed since the development of enzyme replacement therapy, with a shift from purely descriptive studies to interventional research and the exploration of potentially curative approaches. While significant challenges remain, particularly regarding access and affordability, the scientific momentum and investment in rare disease research provide hope for continued advances in understanding and treating this once devastating disorder.
12. Interesting Facts & Lesser-Known Insights
Unique Scientific Aspects
Counterintuitive Laboratory Profile:
- HPP is one of the few conditions where low alkaline phosphatase is pathological
- Most bone diseases present with elevated ALP, making HPP an important diagnostic consideration when ALP is unexpectedly low
- This often leads to missed diagnoses, as clinicians typically focus on elevated rather than decreased values
Evolutionary Significance:
- The ALPL gene is highly conserved across species, indicating its fundamental importance
- Alkaline phosphatase has ancient origins, with similar enzymes found in bacteria
- TNSALP evolved from bacterial phosphatases approximately 100 million years ago
- Some theorize that efficient bone mineralization enabled by TNSALP was crucial for vertebrate terrestrial adaptation
Biochemical Peculiarities:
- HPP exemplifies how accumulation of a substrate (pyrophosphate) can be more harmful than the absence of a product (phosphate)
- The disease demonstrates the delicate balance needed in mineralization – both too much and too little mineralization are problematic
- The same enzyme (TNSALP) plays critical roles in seemingly unrelated processes: bone mineralization, vitamin B6 metabolism, and neural function
Therapeutic Innovation Significance:
- Asfotase alfa represents one of the first successful applications of “bone-targeting” technology in medicine
- The fusion protein includes a deca-aspartate sequence that specifically binds to hydroxyapatite
- This targeted approach has become a model for other bone-directed therapies
Historical and Cultural Perspectives
Historical Cases:
- Retrospective analysis suggests some historical “rickets” cases were likely undiagnosed HPP
- Archaeological evidence from ancient skeletal remains has identified probable cases of severe HPP
- Some folkloric descriptions of “soft-boned” children may have represented HPP
Naming Etymology:
- The term “hypophosphatasia” is somewhat misleading, as phosphate levels are normal or elevated
- The name refers to low alkaline phosphatase (“phosphatase”) activity rather than low phosphate
- This naming convention has contributed to confusion with hypophosphatemia (low phosphate levels)
Treatment Development Story:
- The development of asfotase alfa represents one of the most dramatic therapeutic successes in rare disease history
- Early attempts at enzyme replacement failed because the enzyme didn’t reach bone effectively
- The bone-targeting innovation came from studying how proteins naturally bind to hydroxyapatite
- Children once expected to die are now growing into adulthood, creating new challenges in transition care
Diagnostic Journey:
- The average patient with adult-onset HPP sees 5-7 healthcare providers before diagnosis
- Dental providers often recognize the condition first in milder cases
- Laboratory software often flags low ALP as “normal” since high values are typically of greater concern
- Some patients report being told their low ALP was a sign of exceptional health
Unusual Clinical Manifestations
Paradoxical Calcification Patterns:
- Despite being a disorder of hypomineralization, HPP patients can develop ectopic calcifications
- Particularly in tendons, ligaments, and joints
- This seeming contradiction illustrates the complex role of TNSALP in regulating tissue-specific mineralization
Vitamin B6-Responsive Seizures:
- Seizures in HPP respond to vitamin B6 supplementation but through a different mechanism than classical B6-dependent seizures
- The seizures result not from B6 deficiency but from inability to transport B6 into neurons due to elevated extracellular PLP
- This has provided insights into neuronal vitamin B6 metabolism
Dental Anomalies:
- Premature loss of teeth with intact roots is pathognomonic
- The cementum (connecting teeth to periodontal ligament) is specifically affected
- Dental x-rays often show enlarged pulp chambers and root canals
- Some patients lose only specific teeth, creating unusual dental patterns
Craniosynostosis Mechanism:
- The premature fusion of skull sutures occurs through a different mechanism than in other craniosynostosis syndromes
- Rather than accelerated ossification, it results from abnormal mineral metabolism at suture sites
- This has provided insights into normal cranial development
Myths and Misconceptions
Myth: HPP is a form of rickets or osteomalacia caused by vitamin D deficiency. Fact: Unlike nutritional rickets, HPP involves normal vitamin D metabolism and calcium/phosphate levels. Vitamin D supplementation doesn’t help and can worsen hypercalcemia.
Myth: Low alkaline phosphatase is always benign or beneficial. Fact: While slightly low ALP may be insignificant, persistently low levels warrant investigation for HPP, especially with bone or dental issues.
Myth: All forms of HPP are severe and life-threatening. Fact: HPP spans a wide spectrum from lethal perinatal forms to mild adult-onset disease with primarily dental manifestations.
Myth: Bisphosphonates are appropriate treatments for HPP-related osteoporosis. Fact: Bisphosphonates are contraindicated in HPP as they mimic pyrophosphate and can worsen mineralization defects.
Myth: HPP is only a bone and tooth disease. Fact: TNSALP functions throughout the body, and HPP can affect multiple systems including neurological, respiratory, renal, and muscular systems.
Practical Insights for Patients and Clinicians
Diagnostic Clues:
- Always check age-specific reference ranges for ALP
- Consider HPP in any patient with premature tooth loss, especially with intact roots
- Low or low-normal ALP with unexplained fractures warrants investigation
- Family history of early tooth loss or “soft bones” provides important context
Management Pearls:
- Vitamin B6 supplementation may control seizures when anticonvulsants fail
- Craniosynostosis requires early recognition and neurosurgical evaluation
- Dental implants can be successful despite mineralization defects
- Regular ophthalmological exams important due to risk of craniosynostosis-related vision issues
Quality of Life Considerations:
- Pain management is often challenging and requires multimodal approaches
- Fatigue is a common but underrecognized symptom
- Psychological impact of chronic, rare disease significant
- Patient support groups provide valuable peer connections and resources
Emerging Research Participation:
- Multiple clinical trials recruiting patients with all forms of HPP
- Natural history studies seeking participants across the disease spectrum
- Biobanking initiatives collecting samples for future research
- Patient-driven research priorities increasingly shaping research agendas
Socioeconomic and Global Aspects
Treatment Access Disparities:
- Dramatic inequities in access to enzyme replacement therapy
- Cost per patient can exceed $1 million annually
- Limited availability in many countries
- Various compassionate use and patient assistance programs
Diagnostic Disparities:
- Significant underdiagnosis in regions with limited healthcare
- Laboratory capabilities for specialized testing vary globally
- Genetic testing accessibility highly variable
- Telemedicine consultations with specialists increasing access
Economic Impact:
- Lifetime cost of severe HPP estimated at $5-10 million USD
- Indirect costs from lost productivity, caregiving burden substantial
- Cost-effectiveness analyses of ERT ongoing
- Orphan drug policies critical for treatment development
Advocacy Impact:
- Patient advocacy organizations key to research funding and awareness
- Successful lobbying for insurance coverage in some regions
- Collaboration between patient groups and researchers accelerating progress
- Global networks connecting isolated patients with rare disease
Hypophosphatasia represents a fascinating intersection of genetics, biochemistry, mineralization biology, enzyme targeting, and rare disease medicine. From its counterintuitive laboratory profile to its dramatic treatment success story, HPP offers valuable lessons for clinicians, researchers, and the broader medical community. As understanding continues to evolve and new treatments emerge, the narrative of this once devastating disease continues to transform into one of hope and scientific progress.