
⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
What is Idiopathic Thrombocytopenic Purpura?
Idiopathic Thrombocytopenic Purpura (ITP), now more commonly known as Immune Thrombocytopenia, is an autoimmune bleeding disorder characterized by abnormally low levels of platelets (thrombocytes) in the blood. The term has evolved from “idiopathic” (of unknown cause) to “immune” as research has clarified that ITP results from an immune-mediated process where the body’s immune system mistakenly attacks and destroys platelets.
The condition is defined by a blood platelet count below 100,000 per microliter (normal range: 150,000-450,000/μL) in the absence of other identifiable causes of thrombocytopenia. When platelet counts fall below 20,000/μL, the risk of bleeding significantly increases, and counts below 10,000/μL can lead to life-threatening hemorrhage.
Affected Body Parts/Organs
ITP primarily affects:
- Blood: Specifically platelets, the small cell fragments responsible for blood clotting
- Spleen: Where platelet destruction predominantly occurs
- Bone Marrow: The site of platelet production (megakaryocytes), which may also be affected
- Skin and Mucous Membranes: Where most bleeding manifestations appear
- Immune System: The source of autoantibodies that target platelets
Prevalence and Significance
ITP is a relatively rare condition with an age-adjusted prevalence of approximately 9.5 cases per 100,000 persons in the United States. The annual incidence is estimated to be 2-5 cases per 100,000 children and 2-3 cases per 100,000 adults.
The condition manifests differently in children and adults:
- In children: ITP is often acute and self-limiting, typically following a viral infection, with 80-90% of cases resolving spontaneously within 6 months.
- In adults: ITP tends to be chronic (persisting for more than 12 months), with only about one-third of cases achieving spontaneous remission.
While ITP can affect individuals of any age, there are distinct patterns of incidence:
- Childhood ITP: Peak incidence at ages 2-4 years, with an equal distribution between males and females
- Adult ITP: Peak incidence at ages 20-50 years, with a female-to-male ratio of approximately 2-3:1
The significance of ITP lies in its impact on quality of life, potential for serious bleeding complications, and the need for long-term management in chronic cases. While mortality is generally low (less than 1% in children and about 5% in adults), ITP can cause significant morbidity, particularly in elderly patients and those with severe thrombocytopenia.
History & Discoveries
First Identification
The history of ITP spans several centuries, with early descriptions of purpura (bleeding into the skin) dating back to ancient times. However, the formal recognition and characterization of ITP as a distinct clinical entity evolved gradually:
11th Century: The Persian physician Avicenna (Ibn Sina) provided one of the earliest documented descriptions of a chronic purpura condition that could fit the diagnosis of ITP in his work “The Canon of Medicine.”
16th Century (1556): Portuguese physician Amatus Lusitanus described a boy who developed dark skin macules and bloody discharges for several days without fever, who eventually recovered spontaneously.
17th Century (1658): Lazarus de la Rivière, physician to the King of France, documented similar cases.
18th Century (1735): The German physician and poet Paul Gottlieb Werlhof wrote the most complete initial clinical description of what would later be called “Werlhof’s disease” or “morbus maculosus hemorrhagicus” (spotted hemorrhagic disease).
Key Discoverers
Several key figures contributed to our understanding of ITP:
Paul Kaznelson (1916): While still a medical student in Prague, Kaznelson hypothesized that platelets were being excessively destroyed in the spleen. He convinced his professor to perform the first splenectomy for ITP, which successfully treated the condition. This became the standard treatment for refractory ITP for more than 30 years.
William J. Harrington and James W. Hollingsworth (1950): These researchers conducted a pivotal experiment that laid the groundwork for understanding the immune-mediated platelet destruction in ITP. In what became known as the “Harrington-Hollingsworth Experiment,” Harrington injected himself with blood from an ITP patient and subsequently developed severe thrombocytopenia, demonstrating that ITP was caused by a transferable factor in the plasma (later identified as antiplatelet antibodies) rather than a defect in platelet production.
Major Breakthroughs
The understanding and treatment of ITP have evolved significantly through several key breakthroughs:
Late 19th Century: The identification of platelets as distinct blood components by Giulio Bizzozero (1882) was crucial for later understanding platelet disorders including ITP.
1950s: The Harrington-Hollingsworth experiment definitively established ITP as an immune-mediated disorder, shifting focus from “idiopathic” to “immune” thrombocytopenia.
1960s-1970s: Development of steroid therapy as a first-line treatment for ITP, providing an alternative to splenectomy.
1980s: Introduction of intravenous immunoglobulin (IVIG) as a rapid treatment for severe ITP, especially important for managing acute bleeding episodes.
1990s: Improved understanding of the role of platelet production deficiency in ITP pathophysiology, not just increased destruction.
2000s: Development and approval of thrombopoietin receptor agonists (TPO-RAs), including romiplostim (2008) and eltrombopag (2008), marking a paradigm shift in treatment by stimulating platelet production rather than simply suppressing the immune system.
2010-present: Development of targeted therapies such as rituximab (anti-CD20 antibody) for treatment-resistant cases and advances in understanding the complex immune mechanisms involved in ITP.
Evolution of Medical Understanding
The medical understanding of ITP has evolved dramatically over time:
Initial Recognition (18th-19th centuries): ITP was initially recognized purely as a clinical syndrome of bleeding and purpura without understanding of its cause.
Platelet Connection (late 19th-early 20th centuries): After the discovery of platelets, ITP was linked to low platelet counts, but the mechanism remained unknown.
Bone Marrow Failure Theory (early 20th century): Initially thought to be due to inadequate platelet production in the bone marrow.
Immune-Mediated Destruction (1950s-1960s): The Harrington experiment established that ITP was caused by immune-mediated platelet destruction, not a production defect.
Dual Mechanism Understanding (1980s-present): Recognition that ITP involves both increased platelet destruction and impaired platelet production.
Molecular Era (1990s-present): Identification of specific autoantibodies targeting platelet glycoproteins (particularly GPIIb/IIIa and GPIb/IX) and deeper understanding of the complex immune dysregulation.
Modern Integrated View (21st century): Current understanding recognizes ITP as a heterogeneous disorder with multiple immunopathological mechanisms, including not just autoantibodies but also T-cell mediated cytotoxicity, complement activation, and impaired thrombopoiesis.
This evolution in understanding has directly informed treatment approaches, moving from empirical treatments (splenectomy) to targeted interventions addressing specific aspects of the disease pathophysiology.
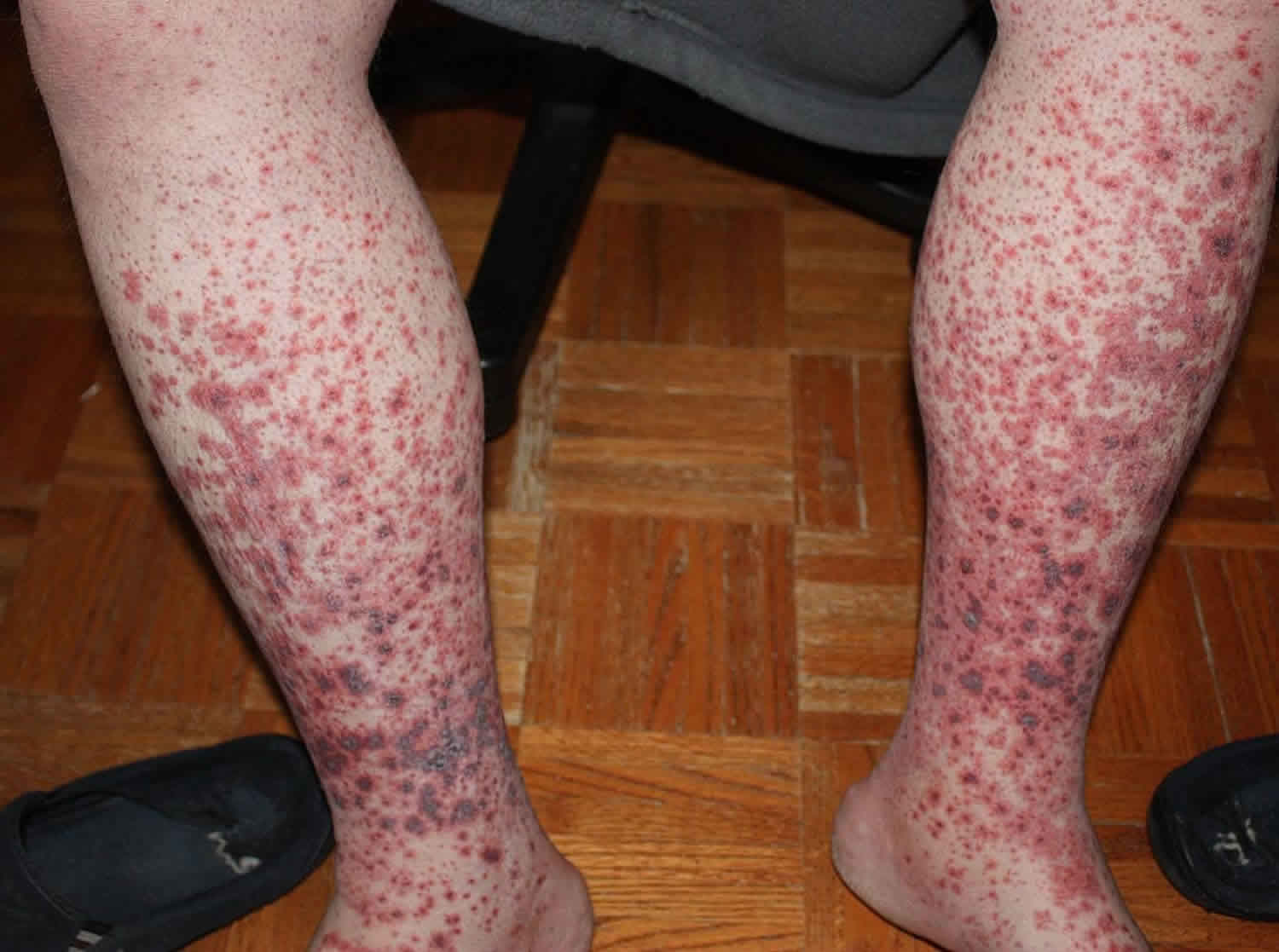
Symptoms
Early Symptoms vs. Advanced-Stage Symptoms
ITP presents with a wide spectrum of clinical manifestations, ranging from incidental findings on a routine blood count to life-threatening hemorrhage. The severity of symptoms generally correlates with the degree of thrombocytopenia.
Early Symptoms (Platelet count 30,000-100,000/μL):
- Often asymptomatic; may be discovered incidentally on routine blood tests
- Mild easy bruising
- Prolonged bleeding from minor cuts or wounds
- Occasional petechiae (tiny red or purple dots on the skin)
- Menorrhagia (heavy menstrual bleeding) in females may be the only presenting symptom
Advanced-Stage Symptoms (Platelet count <30,000/μL):
- Extensive petechiae and purpura (purple patches on the skin)
- Frequent and severe bruising with minimal or no trauma
- Prolonged bleeding from wounds, dental procedures, or surgeries
- Mucosal bleeding (gums, nose, urinary tract)
- Blood blisters in the mouth
Severe Symptoms (Platelet count <10,000/μL):
- Spontaneous mucosal bleeding from multiple sites
- Hematuria (blood in urine)
- Gastrointestinal bleeding
- Retinal hemorrhage
- Intracranial hemorrhage (rare but life-threatening)
- Hemorrhagic bullae (blood-filled blisters) in the mouth or skin
Common vs. Rare Symptoms
Common Symptoms (occurring in >25% of cases):
- Petechiae (pinpoint red/purple spots on skin)
- Purpura (larger patches of purple discoloration)
- Easy bruising
- Prolonged bleeding from minor wounds
- Epistaxis (nosebleeds)
- Gingival bleeding (bleeding gums)
- Fatigue and malaise (possibly related to anemia if bleeding is significant)
Less Common Symptoms (5-25% of cases):
- Menorrhagia (heavy menstrual bleeding)
- Hematuria (blood in urine)
- Blood blisters in the mouth
- Prolonged bleeding after surgery or dental procedures
- Conjunctival hemorrhage (bleeding in the eye)
Rare Symptoms (<5% of cases):
- Gastrointestinal bleeding
- Hemoptysis (coughing up blood)
- Retinal hemorrhage
- Intracranial hemorrhage (bleeding in the brain) – occurs in <1% of children and ~1.5% of adults with ITP
- Visceral hematomas (internal bleeding in organs)
Symptom Progression Over Time
The progression of symptoms in ITP varies significantly between children and adults and between acute and chronic forms of the disease:
In Children (Acute ITP):
- Initial Presentation: Often abrupt onset of petechiae and bruising, typically 1-4 weeks after a viral illness
- Peak Severity: Usually within the first week of diagnosis
- Resolution Phase: In 80-90% of children, symptoms resolve spontaneously within 6 months
- Chronic Phase: In approximately 10-20% of children, symptoms persist beyond 12 months, requiring ongoing management
In Adults (Typically Chronic ITP):
- Initial Presentation: Generally more insidious onset, often discovered incidentally or with mild symptoms
- Fluctuating Course: Symptom severity may wax and wane over months to years
- Exacerbations: May be triggered by infections, medications, or stress
- Stability: Most adults with chronic ITP reach a relatively stable platelet count with appropriate treatment
- Remission: Spontaneous remission occurs in about one-third of adults, while others require ongoing management
Factors Affecting Symptom Progression:
- Age: Older patients generally have more severe bleeding manifestations
- Comorbidities: Conditions like hypertension increase the risk of severe bleeding, particularly intracranial hemorrhage
- Medications: Anticoagulants or antiplatelet drugs can exacerbate bleeding symptoms
- Treatment Response: Individual response to therapy significantly impacts symptom trajectory
- Disease Subtype: Some patients have more refractory disease with persistent severe thrombocytopenia despite multiple treatments
Causes
Biological and Environmental Causes
ITP is an autoimmune disorder where the immune system mistakenly attacks and destroys platelets. The complex pathophysiology involves several mechanisms:
Primary Biological Mechanisms:
Autoantibody Production:
- Production of IgG autoantibodies against platelet membrane glycoproteins (primarily GPIIb/IIIa and GPIb/IX)
- These antibody-coated platelets are recognized and destroyed by macrophages in the spleen and liver through Fc receptor-mediated phagocytosis
- In approximately 60-70% of ITP patients, these autoantibodies can be detected
T-Cell Mediated Platelet Destruction:
- Cytotoxic CD8+ T cells directly attack platelets
- This mechanism is particularly important in patients without detectable autoantibodies
Impaired Platelet Production:
- Autoantibodies also target megakaryocytes (platelet precursors) in the bone marrow
- This leads to reduced platelet production, compounding the problem of increased destruction
- Studies show that thrombopoietin levels are not appropriately elevated in ITP, suggesting dysregulation of the platelet production feedback mechanism
Environmental Triggers:
While ITP is primarily an autoimmune disorder, several environmental factors can trigger its onset:
Viral Infections:
- In children: Common viral infections (measles, chickenpox, rubella, Epstein-Barr virus, influenza)
- In adults: HIV, hepatitis C, cytomegalovirus, Helicobacter pylori
- Molecular mimicry between viral antigens and platelet proteins may initiate the autoimmune response
Medications:
- Certain drugs can induce immune thrombocytopenia through hapten-dependent antibody formation
- Common culprits include quinine, quinidine, sulfonamides, vancomycin, and heparin
- Some vaccines have been rarely associated with transient ITP, particularly MMR vaccine
Environmental Toxins:
- Heavy metals and certain pesticides have been linked to autoimmune disorders, including ITP
- However, direct causative relationships are difficult to establish
Pregnancy:
- Pregnancy can trigger ITP or exacerbate pre-existing ITP
- Hormonal changes and immune system adaptations during pregnancy may play a role
Genetic and Hereditary Factors
While ITP is not primarily a genetic disorder, there is evidence for genetic contributions to susceptibility:
HLA Associations:
- Certain HLA (Human Leukocyte Antigen) types have been associated with increased ITP risk
- HLA-DRB1*0410 has been linked to ITP in some populations
- HLA-DRB10401 and HLA-DRB11501 have been associated with chronic ITP
Immune Regulation Genes:
- Polymorphisms in genes regulating immune function (such as FCGR, TNFAIP3, and PTPN22) have been associated with ITP susceptibility
- Specifically, the FCGR3a-V158 allele and KIRDS2/DL2 have been identified as increasing susceptibility to ITP
Familial Clustering:
- While rare, familial cases of ITP have been reported
- First-degree relatives of ITP patients have a slightly higher risk of developing autoimmune disorders, including ITP
- This suggests shared genetic risk factors for autoimmunity
Protective Genetic Factors:
- The KIR2DS5 gene has been identified as potentially protective against ITP development
It’s important to note that ITP is not considered a hereditary disease in the classical sense. Rather, individuals may inherit a genetic predisposition to autoimmunity, which combined with environmental triggers, may lead to ITP.
Known Triggers or Exposure Risks
Several specific triggers and exposure risks have been associated with the onset or exacerbation of ITP:
Infections:
- Viral: Particularly important in childhood ITP, with approximately 60% of cases following a viral illness
- Bacterial: Helicobacter pylori infection has been linked to ITP in adults, with eradication leading to improvement in some cases
- HIV and HCV: Both are well-established causes of secondary ITP
Vaccines:
- Measles-Mumps-Rubella (MMR) vaccine has been associated with a small increased risk of ITP (1 case per 40,000 vaccinations)
- This risk is significantly lower than the risk of ITP from natural measles infection
- The ITP that follows vaccination is usually transient and resolves within 6 months
Medications:
- Drug-induced thrombocytopenia can mimic ITP
- Common culprits include heparin, quinine/quinidine, trimethoprim-sulfamethoxazole, vancomycin, and glycoprotein IIb/IIIa inhibitors
- The mechanism typically involves drug-dependent antibodies that bind to platelets only in the presence of the drug
Pregnancy:
- Up to 5% of ITP cases are associated with pregnancy
- Typically presents in the first or early second trimester
- May recur in subsequent pregnancies
Surgery and Trauma:
- Major surgery or trauma can sometimes trigger the onset of ITP in predisposed individuals
- The mechanism likely involves inflammation and immune system activation
Underlying Conditions:
- Other autoimmune disorders: Systemic lupus erythematosus, antiphospholipid syndrome, autoimmune thyroid disease
- Lymphoproliferative disorders: Chronic lymphocytic leukemia, lymphoma
- Immunodeficiency states: Common variable immunodeficiency
Psychological Stress:
- Some studies suggest that severe psychological stress may trigger or worsen ITP in some patients
- The mechanism likely involves stress-induced immune dysregulation
Understanding these triggers is crucial for differentiating primary (idiopathic) ITP from secondary ITP, which occurs as a result of an underlying condition. Management approaches may differ based on whether a specific trigger can be identified and addressed.
Risk Factors
Who is Most at Risk (Age, Gender, Occupation, Lifestyle)
ITP affects distinct demographic groups with varying patterns:
Age-Related Risk:
- Children: Peak incidence at ages 2-4 years
- Adults: Highest incidence between 20-50 years
- Elderly: Increasing incidence after age 60, with different clinical characteristics and greater risk of severe bleeding
Gender Distribution:
- Children: Nearly equal distribution between males and females
- Adults: Strong female predominance (female:male ratio of 2-3:1)
- Elderly (>60 years): Gender distribution becomes more equal again
Occupational Risks: While ITP itself is not directly linked to specific occupations, certain professions may carry increased risks or complications:
- Healthcare workers with exposure to bloodborne pathogens (higher risk of secondary ITP due to hepatitis C or HIV exposure)
- Individuals working in jobs with high bleeding risk (e.g., construction, forestry, sports) face greater complications if ITP develops
- Workers exposed to certain chemicals or toxins that may trigger autoimmune responses
Lifestyle Factors:
- Poor nutrition (particularly vitamin deficiencies) may exacerbate symptoms
- High stress levels may trigger or worsen autoimmune conditions, including ITP
- Alcohol consumption can worsen thrombocytopenia and increase bleeding risk
- Smoking has been associated with poorer treatment response
Environmental, Occupational, and Genetic Factors
Environmental Factors:
- Geographic Variations: Some studies suggest geographical differences in ITP prevalence, potentially related to infectious disease patterns, environmental toxins, or genetic population differences
- Seasonal Patterns: Higher incidence of childhood ITP during winter and spring months, correlating with peaks in viral infections
- Socioeconomic Status: Limited evidence suggests potential associations with access to healthcare and environmental exposures
Occupational Exposures:
- Chemical Exposures: Pesticides, organic solvents, and certain industrial chemicals have been linked to autoimmune disorders
- Radiation Exposure: Ionizing radiation may contribute to immune dysregulation
- Shift Work: Disruption of circadian rhythms may impact immune function
Genetic Susceptibility Factors:
- HLA Types: Specific HLA haplotypes show associations with ITP risk and chronicity
- Fc Receptor Polymorphisms: Variations in Fc gamma receptor genes affect platelet clearance
- Immune Regulation Genes: Polymorphisms in genes controlling immune tolerance and regulation
- Family History: Slightly increased risk among those with first-degree relatives with autoimmune disorders
Impact of Pre-existing Conditions
Several pre-existing conditions can significantly influence ITP risk, presentation, and management:
Autoimmune Disorders:
- Systemic Lupus Erythematosus (SLE): 10-15% of SLE patients develop secondary ITP
- Antiphospholipid Syndrome: Often associated with thrombocytopenia and increased thrombotic risk
- Autoimmune Thyroid Diseases: Higher prevalence of ITP in patients with Hashimoto’s thyroiditis or Graves’ disease
- Rheumatoid Arthritis: Shares some immunopathological mechanisms with ITP
Infectious Diseases:
- HIV Infection: Approximately 10-20% of HIV patients develop thrombocytopenia
- Hepatitis C Virus: Associated with thrombocytopenia through multiple mechanisms
- Helicobacter pylori: Eradication can lead to platelet count improvements in some ITP patients
Hematologic Conditions:
- Lymphoproliferative Disorders: Chronic lymphocytic leukemia and lymphomas can cause secondary ITP
- Myelodysplastic Syndromes: Can present with isolated thrombocytopenia mimicking ITP, particularly in elderly patients
- Evans Syndrome: Combined autoimmune thrombocytopenia and hemolytic anemia
Other Medical Conditions:
- Liver Disease: Cirrhosis can cause thrombocytopenia through multiple mechanisms
- Pregnancy: Can trigger or exacerbate ITP
- Immunodeficiency States: Common variable immunodeficiency and IgA deficiency have associations with ITP
- Post-Transplantation: Solid organ or bone marrow transplant recipients have increased risk
Medication Use:
- Anticoagulants: Increase bleeding risk in patients with ITP
- Immunosuppressive Drugs: May either mask or trigger ITP, depending on the mechanism
- Certain Antibiotics and Anticonvulsants: Can cause drug-induced thrombocytopenia
Understanding these risk factors is essential for early identification of high-risk individuals, appropriate diagnostic evaluation, and optimized management strategies to prevent complications.
Complications
Complications Arising from ITP
ITP can lead to various complications, ranging from mild to life-threatening:
Bleeding Complications:
- Mucosal Bleeding: Frequent nosebleeds, gingival bleeding, or excessive menstrual bleeding
- Gastrointestinal Hemorrhage: Bleeding in the digestive tract, which can be occult or overt
- Genitourinary Bleeding: Hematuria (blood in urine) or excessive menstrual bleeding in females
- Retinal Hemorrhage: Bleeding in the retina that can affect vision
- Intracranial Hemorrhage (ICH): The most feared complication, occurring in approximately 0.5-1% of children and 1.5-3% of adults with severe ITP
- Risk factors for ICH include: platelet counts <10,000/μL, head trauma, hypertension, previous bleeding episodes, and advanced age
- ICH is associated with a mortality rate of approximately 40-50%
Treatment-Related Complications:
- Corticosteroid Complications: Weight gain, diabetes, osteoporosis, avascular necrosis, mood disorders, increased infection risk, hypertension, and skin changes
- IVIG-Related Complications: Headache, aseptic meningitis, thrombosis, renal insufficiency, and allergic reactions
- Splenectomy Complications: Surgical risks, overwhelming post-splenectomy infection (OPSI), thrombosis, and pulmonary hypertension
- Thrombopoietin Receptor Agonist Complications: Thrombosis, rebound thrombocytopenia, bone marrow fibrosis, and hepatotoxicity
- Rituximab Complications: Infusion reactions, progressive multifocal leukoencephalopathy (very rare), hepatitis B reactivation, and hypogammaglobulinemia
Psychosocial Complications:
- Reduced quality of life
- Anxiety and depression
- Limitations in activities and lifestyle
- Employment difficulties
- Financial burden of chronic illness
Long-term Impact on Organs and Overall Health
ITP and its treatments can have significant long-term impacts on multiple organ systems:
Hematologic System:
- Chronic anemia from recurrent bleeding
- Potential for iron deficiency from chronic blood loss
- Rarely, secondary malignancies associated with immunosuppressive therapy
Cardiovascular System:
- Paradoxically, despite bleeding tendencies, ITP is associated with an increased risk of thrombotic events
- The risk of thrombosis is 1.5-2 times higher in ITP patients compared to the general population
- This risk is further increased with certain treatments, particularly splenectomy and thrombopoietin receptor agonists
Immune System:
- Long-term immunosuppression from treatments can increase infection risk
- Post-splenectomy patients have lifelong increased susceptibility to encapsulated bacterial infections
- Some treatments may increase the risk of secondary autoimmune disorders
Musculoskeletal System:
- Osteoporosis and fractures from prolonged corticosteroid use
- Avascular necrosis, particularly affecting the femoral head
- Muscle weakness and atrophy
Endocrine System:
- Adrenal suppression from chronic steroid therapy
- Metabolic abnormalities including glucose intolerance and diabetes
- Weight gain and body composition changes
Neuropsychiatric Health:
- Mood disorders, cognitive changes, and sleep disturbances from treatments
- Potential neurocognitive sequelae in survivors of intracranial hemorrhage
- Reduced quality of life and psychosocial well-being
Reproductive Health:
- Menstrual irregularities and fertility concerns in females
- Pregnancy complications including increased maternal bleeding risk and fetal thrombocytopenia
- Some treatments may affect fertility or are contraindicated during pregnancy
Potential Disability or Fatality Rates
Disability:
- Chronic ITP can lead to significant disability in approximately 20-30% of patients
- Major sources of disability include:
- Activity restrictions due to bleeding risk
- Neurological deficits following intracranial hemorrhage
- Complications of treatment (particularly long-term corticosteroid use or post-splenectomy complications)
- Chronic fatigue, which affects up to 40% of ITP patients regardless of platelet count
- Studies show that health-related quality of life is significantly reduced in adults with chronic ITP, comparable to other chronic diseases like rheumatoid arthritis
Mortality:
- Overall mortality rates in ITP have decreased significantly with modern management
- Current mortality rates:
- Children: <1% (most deaths from intracranial hemorrhage)
- Adults: approximately 5% (higher in elderly patients)
- Elderly patients (>60 years) with severe thrombocytopenia: 5-year mortality up to 47.8%
- Causes of death in ITP patients:
- Hemorrhage: Primarily intracranial and gastrointestinal bleeding
- Infections: Particularly in immunosuppressed patients and post-splenectomy
- Thrombotic events: Including myocardial infarction, stroke, and pulmonary embolism
- Treatment complications: Surgery-related mortality, drug-induced complications
- Comorbidities: Particularly in elderly patients
The mortality rate is significantly influenced by:
- Age (higher in patients >60 years)
- Platelet count (higher risk with counts <10,000/μL)
- Previous bleeding history
- Comorbidities (particularly cardiovascular disease)
- Response to treatment
With appropriate management, most patients with ITP can live normal lifespans with good quality of life, though chronic disease management may be necessary.
Diagnosis & Testing
Common Diagnostic Procedures
The diagnosis of ITP is primarily one of exclusion, requiring a systematic approach to rule out other causes of thrombocytopenia. Common diagnostic procedures include:
Clinical History and Physical Examination:
- Detailed bleeding history (onset, severity, location, precipitating factors)
- Medication history (to identify drug-induced thrombocytopenia)
- Family history (to exclude hereditary thrombocytopenias)
- Recent infections or vaccinations
- Symptoms of underlying systemic disorders
- Physical examination focusing on:
- Sites and extent of bleeding (petechiae, purpura, bruising)
- Lymphadenopathy or hepatosplenomegaly (suggestive of lymphoproliferative disorders or infections)
- Signs of systemic autoimmune diseases
Laboratory Testing:
- Complete Blood Count (CBC): The cornerstone of ITP diagnosis
- Isolated thrombocytopenia (platelets <100,000/μL) with otherwise normal blood counts
- Normal hemoglobin and white blood cell count (unless there has been significant bleeding)
- Peripheral Blood Smear: Essential to confirm thrombocytopenia and exclude other disorders
- Reduced number of platelets
- Normal platelet morphology (occasionally larger platelets)
- Absence of schistocytes, blasts, or other abnormal cells
- Basic Coagulation Studies:
- Prothrombin time (PT) and activated partial thromboplastin time (aPTT) are typically normal
- Abnormal results suggest an alternative diagnosis or coexisting coagulopathy
Additional Diagnostic Tests: Based on clinical presentation and initial findings, additional tests may include:
- Immunological Testing:
- Antinuclear antibody (ANA) and anti-double-stranded DNA (anti-dsDNA) to screen for lupus
- Antiphospholipid antibodies
- Direct antiglobulin test (DAT) to rule out Evans syndrome
- Infectious Disease Screening:
- HIV testing
- Hepatitis C testing
- Helicobacter pylori testing in appropriate populations
- Bone Marrow Examination: Not routinely required but indicated in certain scenarios (see below)
Medical Tests (Blood Tests, Imaging, Biopsies)
Blood Tests:
Standard Tests for ITP Diagnosis:
- Complete blood count (CBC) with differential
- Peripheral blood smear examination
- Reticulocyte count
- Basic metabolic panel
- Liver function tests
- Coagulation studies (PT, aPTT, fibrinogen)
- Blood type and Rh status (relevant for anti-D therapy)
Specialized Blood Tests (based on clinical indication):
- Autoantibody Testing: Anti-platelet antibody testing has limited utility due to variable sensitivity (30-60%) and specificity
- Immunoglobulin Levels: To exclude common variable immunodeficiency
- Thrombopoietin Levels: May be helpful in distinguishing ITP from other causes of thrombocytopenia
- Glycoprotein-Specific Antibody Testing: More specific than general antiplatelet antibodies but limited availability
- Flow Cytometric Analysis: For reticulated platelets (young platelets) – increased in ITP
Imaging Studies:
- Imaging is not routinely required for ITP diagnosis but may be indicated in specific situations:
- Abdominal Ultrasound: To assess spleen size and rule out splenic sequestration
- CT Scan or MRI: In cases of suspected intracranial hemorrhage
- Chest X-ray: To exclude mediastinal masses in suspected lymphoma
- PET-CT: May be indicated if there is concern for underlying malignancy
Bone Marrow Examination:
- Not required for typical cases but indicated in:
- Patients >60 years of age (to exclude myelodysplastic syndrome or leukemia)
- Atypical features (abnormalities in other blood cell lines)
- Prior to certain treatments (e.g., TPO-RA therapy in some cases)
- Failure to respond to first-line therapy
- Consideration of aplastic anemia or other bone marrow failure syndrome
The bone marrow examination in ITP typically shows:
- Normal to increased number of megakaryocytes (platelet precursors)
- Normal erythroid and myeloid precursors
- Absence of fibrosis, dysplasia, or infiltration by abnormal cells
Other Specialized Tests:
- Platelet Survival Studies: Rarely performed but can demonstrate shortened platelet lifespan
- HLA Typing: In preparation for certain treatments or transplantation
- Genetic Testing: To exclude hereditary thrombocytopenias in suspected cases
- Molecular Testing: For specific mutations in atypical cases or when considering myeloid neoplasms
Early Detection Methods and Their Effectiveness
Early detection of ITP is challenging due to its nature as a diagnosis of exclusion and the absence of specific biomarkers. However, certain approaches can facilitate earlier diagnosis:
Routine Blood Testing:
- Incidental finding of thrombocytopenia on routine CBC is common in asymptomatic ITP
- Regular monitoring in high-risk groups (e.g., patients with other autoimmune disorders, HIV, or hepatitis C)
- Effectiveness: Moderate – detects asymptomatic cases but may lead to overinvestigation
Clinical Vigilance in High-Risk Groups:
- Monitoring for early bleeding signs in patients with risk factors
- Lower threshold for CBC testing in patients with mild bruising or bleeding symptoms
- Effectiveness: Moderate – depends on clinical awareness and access to care
Specialized Screening Methods (research settings):
- Reticulated Platelet Percentage: Higher in ITP than in other causes of thrombocytopenia
- Plasma Glycocalicin Index: Elevated in ITP due to increased platelet destruction
- Thrombopoietin Levels: Lower than expected for the degree of thrombocytopenia in ITP
- Platelet Autoantibody Testing: Limited sensitivity but may help in ambiguous cases
- Effectiveness: Limited – these tests are not widely available and have variable sensitivity/specificity
Effectiveness of Current Diagnostic Approaches:
- Sensitivity: The current diagnostic approach has good sensitivity for detecting thrombocytopenia but limited sensitivity for confirming ITP specifically
- Specificity: Moderate to high when a comprehensive evaluation is performed
- Time to Diagnosis: Variable – can be immediate with clear presentations or delayed in complex cases
- Cost-Effectiveness: The exclusionary approach leads to extensive testing in some cases, reducing cost-effectiveness
Challenges in Early Diagnosis:
- ITP remains a diagnosis of exclusion with no pathognomonic test
- Overlap with other causes of thrombocytopenia
- Variable presentations from asymptomatic to severe bleeding
- Evolving understanding of disease subtypes and mechanisms
Future Directions in Early Detection:
- Development of more specific biomarkers for ITP
- Improved platelet autoantibody testing with higher sensitivity and specificity
- Genomic and proteomic approaches to identify ITP subtypes
- Point-of-care testing for rapid assessment of platelet function and number
Early detection and diagnosis of ITP remain challenging but are critical for preventing severe bleeding complications, particularly in high-risk populations such as the elderly and those with comorbidities.
Treatment Options
Standard Treatment Protocols
Treatment of ITP follows a stratified approach based on bleeding severity, platelet count, patient characteristics, and phase of the disease. Current standard protocols include:
Observation (Watch and Wait):
- For patients with platelet counts >30,000/μL without significant bleeding
- Regular monitoring of platelet counts
- Education regarding bleeding precautions and when to seek medical attention
- Avoidance of medications that affect platelet function (aspirin, NSAIDs)
First-Line Treatments:
Corticosteroids:
- Standard initial therapy for most adult patients with newly diagnosed ITP
- Options include:
- Prednisone (1-2 mg/kg/day for 2-4 weeks, followed by tapering)
- Dexamethasone (40 mg/day for 4 days, repeated if necessary)
- Methylprednisolone (for severe cases requiring hospitalization)
- Expected response in 60-80% of patients, though sustained remission occurs in only 20-30%
Intravenous Immunoglobulin (IVIG):
- Used for rapid increase in platelet count in patients with severe bleeding or requiring urgent surgery
- Typical dose: 1 g/kg/day for 1-2 days
- Rapid response (within 24-48 hours) in approximately 80% of patients
- Effect is temporary (2-4 weeks)
Anti-D Immunoglobulin (for Rh-positive, non-splenectomized patients):
- Alternative to IVIG in appropriate patients
- Typical dose: 50-75 μg/kg as a single dose
- Works by preferentially destroying antibody-coated red blood cells, sparing platelets
- Not available in all countries
Second-Line Treatments (for persistent/chronic ITP or relapse after first-line therapy):
Thrombopoietin Receptor Agonists (TPO-RAs):
- Romiplostim: Weekly subcutaneous injection, 1-10 μg/kg
- Eltrombopag: Daily oral medication, 25-75 mg
- Avatrombopag: Daily oral medication, 20-40 mg
- Response rates of 80-90% with continued therapy
- Now increasingly used earlier in the disease course
Rituximab (anti-CD20 monoclonal antibody):
- Targets B cells responsible for antibody production
- Standard dose: 375 mg/m² weekly for 4 weeks or lower dose regimens
- Response in approximately 60% of patients, with 20-30% maintaining long-term remission
- Off-label use in many countries for ITP
Splenectomy:
- Historically the standard second-line therapy, now less commonly performed
- Long-term response in approximately 60-70% of patients
- Now typically reserved for patients who fail medical therapy
- Can be performed laparoscopically with reduced morbidity
- Lifelong increased risk of infection and thrombosis
Third-Line Treatments (for refractory ITP):
Immunosuppressive Agents:
- Azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine
- Variable response rates (30-60%)
- Significant side effect profiles including increased infection risk
Combination Therapies:
- TPO-RA plus immunosuppressant
- TPO-RA plus rituximab
- Multiple immunosuppressive agents
Experimental Approaches:
- Complement inhibitors
- Syk inhibitors (fostamatinib)
- Neonatal Fc receptor blockers
Emergency Management (for life-threatening bleeding):
- Platelet transfusions (though short-lived due to rapid destruction)
- High-dose IVIG and corticosteroids
- Emergency splenectomy in select cases
- Recombinant Factor VIIa in extreme cases
- Tranexamic acid for mucosal bleeding
Special Populations:
Children:
- Observation alone for many cases of newly diagnosed ITP
- First-line: corticosteroids (shorter course), IVIG, or anti-D
- Second-line: similar to adults but with more caution regarding long-term immunosuppression
Pregnant Women:
- First-line: IVIG or corticosteroids
- TPO-RAs generally avoided due to limited safety data
- Target platelet count >30,000/μL during pregnancy and >50,000/μL for delivery
Elderly Patients:
- Increased caution with corticosteroids due to side effects
- Earlier consideration of TPO-RAs
- Careful assessment of bleeding versus thrombotic risk
Medications, Surgeries, and Therapies
Medications for ITP:
Corticosteroids:
- Mechanism: Reduce antibody production, decrease phagocytosis, and improve vascular integrity
- Specific Agents:
- Prednisone/prednisolone: Standard oral therapy
- Dexamethasone: Higher potency, shorter course
- Methylprednisolone: Intravenous option for severe cases
- Side Effects: Weight gain, diabetes, osteoporosis, hypertension, mood changes, insomnia, increased infection risk, avascular necrosis
Intravenous Immunoglobulin (IVIG):
- Mechanism: Blocks Fc receptors on macrophages, neutralizes autoantibodies, modulates cytokine production
- Dosing: 1 g/kg/day for 1-2 days, or 0.4 g/kg/day for 5 days
- Side Effects: Headache, aseptic meningitis, thrombotic events, renal insufficiency, infusion reactions
Anti-D Immunoglobulin:
- Mechanism: Competitive inhibition of Fc receptors by coating RBCs
- Dosing: 50-75 μg/kg as a single dose
- Side Effects: Hemolysis, headache, fever, chills, rare disseminated intravascular coagulation
Thrombopoietin Receptor Agonists (TPO-RAs):
- Mechanism: Stimulate platelet production by activating the thrombopoietin receptor
- Specific Agents:
- Romiplostim (Nplate): Weekly subcutaneous injection
- Eltrombopag (Promacta/Revolade): Daily oral tablet
- Avatrombopag (Doptelet): Daily oral tablet
- Side Effects: Headache, fatigue, thrombotic events, rebound thrombocytopenia upon discontinuation, rare bone marrow fibrosis
Rituximab:
- Mechanism: Depletes CD20+ B cells, reducing autoantibody production
- Dosing: 375 mg/m² weekly for 4 weeks or lower-dose regimens
- Side Effects: Infusion reactions, increased infection risk, rare progressive multifocal leukoencephalopathy, hepatitis B reactivation
Immunosuppressive Agents:
- Azathioprine: Purine synthesis inhibitor; 1-2 mg/kg/day
- Mycophenolate Mofetil: Inhibits lymphocyte proliferation; 1000 mg twice daily
- Cyclosporine: Calcineurin inhibitor; 2.5-3 mg/kg/day in divided doses
- Cyclophosphamide: Alkylating agent; various dosing regimens
- Side Effects: Cytopenia, increased infection risk, gastrointestinal disturbances, potential for secondary malignancies
Newer Targeted Therapies:
- Fostamatinib (Tavalisse): Spleen tyrosine kinase (Syk) inhibitor; 100-150 mg twice daily
- Side Effects: Diarrhea, hypertension, nausea, dizziness, respiratory infections
Surgical Interventions:
Splenectomy:
- Approach: Laparoscopic (preferred) or open
- Mechanism: Removes the primary site of platelet destruction and antibody production
- Efficacy: 60-70% long-term response rate
- Complications: Surgical risks, overwhelming post-splenectomy infection (OPSI), thrombosis, pulmonary hypertension
- Preoperative Considerations:
- Vaccination against encapsulated organisms (pneumococcus, meningococcus, Haemophilus influenzae)
- Platelet count optimization (typically >50,000/μL)
- Preoperative platelet response predictors (indium-labeled platelet scanning largely replaced by clinical factors)
Accessory Splenectomy:
- For patients who relapse after initial splenectomy and are found to have accessory splenic tissue
- Identified through nuclear medicine studies (heat-damaged red cell scan)
Additional Therapies:
Platelet Transfusions:
- Used only for severe bleeding or before urgent procedures
- Limited efficacy due to rapid destruction of transfused platelets
- Often combined with IVIG to prolong platelet survival
Hemostatic Agents:
- Tranexamic Acid: Antifibrinolytic agent; useful for mucosal bleeding
- Epsilon Aminocaproic Acid: Alternative antifibrinolytic agent
- Recombinant Factor VIIa: For life-threatening bleeding unresponsive to other measures
Plasma Exchange/Plasmapheresis:
- Rarely used; removes autoantibodies from circulation
- Temporary effect; typically followed by immunosuppressive therapy
Stem Cell Transplantation:
- Reserved for severe, refractory cases with significant morbidity
- High-risk procedure with potential for significant complications
- Limited experience in ITP; primarily case reports and small series
Complementary Approaches:
- Vitamin D supplementation (some evidence for immunomodulatory effects)
- H. pylori eradication when present (especially effective in certain populations)
- Lifestyle modifications to reduce bleeding risk
- Psychological support and stress management
Selection of therapy should be individualized based on:
- Severity of thrombocytopenia and bleeding
- Patient age and comorbidities
- Previous treatment history and response
- Patient preferences and quality of life considerations
- Regional availability and cost of therapies
Emerging Treatments and Clinical Trials
The treatment landscape for ITP continues to evolve with numerous innovative approaches in development:
Novel Thrombopoietic Agents:
- Subcutaneous Romiplostim Formulation: Self-administered, prefilled syringes for improved convenience
- Long-Acting TPO Mimetics: Requiring less frequent dosing
- Oral TPO Receptor Agonists: New compounds with improved pharmacokinetics
- Clinical Trial Status: Phase II-III studies underway for several compounds
Targeted Immunomodulatory Approaches:
Syk Inhibitors:
- Fostamatinib: FDA-approved in 2018; inhibits spleen tyrosine kinase, reducing platelet destruction
- Next-Generation Syk Inhibitors: More selective compounds with improved efficacy and reduced side effects
- Clinical Trial Status: Fostamatinib in post-marketing studies; newer compounds in Phase I-II
Bruton’s Tyrosine Kinase (BTK) Inhibitors:
- Mechanism: Disrupts B-cell signaling and antibody production
- Compounds: Ibrutinib, acalabrutinib, rilzabrutinib
- Clinical Trial Status: Phase II-III trials showing promising results, with response rates of 40-60%
Neonatal Fc Receptor (FcRn) Inhibitors:
- Mechanism: Blocks recycling of pathogenic IgG antibodies, accelerating their clearance
- Compounds: Rozanolixizumab, efgartigimod, nipocalimab
- Clinical Trial Status: Phase II-III trials with encouraging early results
Complement Pathway Inhibitors:
- Mechanism: Prevents complement-mediated platelet destruction
- Compounds: Sutimlimab (anti-C1s), pegcetacoplan (anti-C3)
- Clinical Trial Status: Phase I-II trials in refractory ITP
Proteasome Inhibitors:
- Mechanism: Targets plasma cells producing antiplatelet antibodies
- Compounds: Bortezomib, carfilzomib
- Clinical Trial Status: Phase II studies and case series showing efficacy in refractory ITP
Cell-Based Therapies:
Regulatory T-Cell (Treg) Therapy:
- Mechanism: Restores immune tolerance by expanding/infusing regulatory T cells
- Approaches: Low-dose IL-2 therapy, ex vivo expanded Tregs
- Clinical Trial Status: Early-phase clinical trials
Mesenchymal Stem Cell Therapy:
- Mechanism: Immunomodulatory and regenerative effects
- Approaches: Allogeneic or autologous transplantation
- Clinical Trial Status: Phase I-II trials, primarily in refractory cases
Chimeric Antigen Receptor (CAR) T-Cell Therapy:
- Mechanism: Engineered T cells targeting autoreactive B cells
- Clinical Trial Status: Preclinical and early clinical exploration
Novel Drug Delivery Systems:
- Nanoparticle-based immunomodulatory drug delivery
- Controlled-release formulations of existing therapies
- Clinical Trial Status: Primarily preclinical with some Phase I studies
Combination Therapy Approaches:
- TPO-RA + Immunosuppressant: Enhanced efficacy with potential for treatment discontinuation
- Rituximab + TPO-RA: Synergistic effects targeting both production and destruction
- Clinical Trial Status: Several Phase II-III trials evaluating combinations
Disease-Modifying Approaches:
- Early Intensive Therapy: Aiming to induce long-term remission through early aggressive intervention
- Targeted Therapy Based on Patient-Specific Mechanisms: Personalized medicine approach
- Clinical Trial Status: Ongoing studies evaluating early intervention strategies
Notable Recent Clinical Trials:
FIT1 Trial: Fostamatinib in persistent/chronic ITP, demonstrating efficacy in patients who failed previous therapies
FLIGHT Trial: Evaluating mycophenolate mofetil as first-line therapy alongside corticosteroids
RIGEL Study: Examining long-term safety and efficacy of rituximab in ITP
TRUST Study: Investigating treatment-free remission after TPO-RA therapy
AVA-ITP-302 Trial: Phase 3 study of avatrombopag in persistent/chronic ITP
Access to Clinical Trials:
- International clinical trial registries (ClinicalTrials.gov, EU Clinical Trials Register)
- ITP Support Association and patient advocacy groups provide information on trial enrollment
- Referral centers for ITP often participate in clinical trials
The emerging treatments for ITP reflect a shift from non-specific immunosuppression to targeted approaches addressing specific disease mechanisms, with the goals of improved efficacy, reduced side effects, and potential for long-term remission or cure.
Prevention & Precautionary Measures
How ITP Can Be Prevented
Primary prevention of ITP is challenging since it is largely an autoimmune disorder with unclear triggering factors. However, certain strategies can help prevent or mitigate secondary causes and reduce the risk of complications:
Prevention of Secondary ITP:
Infectious Disease Prevention:
- Hepatitis C and HIV testing and prevention strategies
- Helicobacter pylori screening and eradication in high-prevalence areas
- Appropriate vaccination to prevent viral illnesses associated with ITP
Medication Vigilance:
- Careful monitoring when using medications associated with drug-induced thrombocytopenia
- Prompt discontinuation of suspected causative medications
- Awareness of cross-reactive medications in patients with history of drug-induced thrombocytopenia
Management of Underlying Conditions:
- Optimal control of autoimmune diseases that can trigger secondary ITP
- Monitoring of lymphoproliferative disorders for early detection of associated ITP
Prevention of ITP Exacerbation:
- Regular monitoring of platelet counts in those with known ITP
- Adherence to prescribed treatment regimens
- Avoidance of triggers known to worsen individual cases
- Prompt management of infections or other illnesses that may trigger flares
Prevention of Complications:
Bleeding Prevention:
- Maintaining adequate platelet counts through appropriate treatment
- Education regarding bleeding precautions
- Avoidance of physical activities with high risk of trauma during periods of severe thrombocytopenia
- Prompt management of minor bleeding to prevent progression
Thrombosis Prevention:
- Appropriate thromboprophylaxis in high-risk patients, especially post-splenectomy
- Careful monitoring for thrombotic complications with certain treatments (TPO-RAs)
- Management of cardiovascular risk factors
Infection Prevention Post-Splenectomy:
- Appropriate vaccinations before and after splenectomy
- Prophylactic antibiotics as indicated
- Prompt evaluation and treatment of febrile illnesses
Lifestyle Changes and Environmental Precautions
Patients with ITP can adopt various lifestyle modifications and precautions to reduce bleeding risk and improve overall outcomes:
Physical Activity Modifications:
During Severe Thrombocytopenia (platelets <30,000/μL):
- Avoidance of contact sports and high-impact activities
- Modification of exercise routines to reduce injury risk
- Consideration of helmet use for outdoor activities even with moderate risk
- Swimming as a safe exercise option (with proper supervision)
With Moderate Thrombocytopenia (platelets 30,000-50,000/μL):
- Limitation of high-risk activities
- Appropriate protective equipment for recreational activities
- Supervised exercise programs
With Mild Thrombocytopenia or Normal Platelets:
- Gradual return to normal activities as platelet count improves
- Continued caution with extreme sports or high-risk activities
Dietary Considerations:
Nutritional Support:
- Well-balanced diet rich in essential nutrients
- Adequate protein intake to support immune function
- Consideration of iron supplementation if deficient due to blood loss
- Sufficient vitamin D and calcium, especially for patients on corticosteroids
Food-Drug Interactions:
- Avoidance of grapefruit juice with certain medications (e.g., eltrombopag)
- Timing of eltrombopag relative to calcium-containing foods and supplements
- Limited alcohol consumption, particularly with liver-metabolized medications
Complementary Approaches:
- Limited evidence for specific diets affecting ITP course
- Some patients report benefit from anti-inflammatory diets, though without strong scientific validation
Home Safety Measures:
- Removal of trip hazards to prevent falls
- Installation of grab bars in bathrooms
- Non-slip mats in showers and bathtubs
- Proper lighting in all areas
- Padded corners on furniture during periods of severe thrombocytopenia
Workplace Accommodations:
- Modified duties to reduce injury risk in physically demanding jobs
- Accommodations for frequent medical appointments
- Education of coworkers about appropriate emergency response
- Consideration of work-from-home options during periods of severe thrombocytopenia
Travel Considerations:
- Medical identification bracelet or card
- Adequate medication supply
- Information about medical facilities at destination
- Travel insurance with coverage for pre-existing conditions
- Consultation with hematologist before travel to remote areas
- Consideration of altitude effects on patients with significant anemia
Stress Management:
- Stress reduction techniques (meditation, yoga, mindfulness)
- Adequate sleep hygiene
- Psychological support as needed
- Support group participation
Vaccines (if applicable) or Preventive Screenings
While there are no vaccines specifically to prevent ITP, several vaccinations and preventive measures are relevant for ITP management:
Vaccination Considerations in ITP Patients:
General Vaccination Guidelines:
- Most inactivated vaccines are safe for ITP patients
- Live vaccines may be contraindicated during periods of immunosuppression
- Vaccination timing ideally during periods of stable platelet counts
- Consider IVIG or platelet transfusion before vaccination if platelets are very low
Specific Vaccines:
- Pre-Splenectomy Vaccinations (ideally at least 2 weeks before surgery):
- Pneumococcal vaccines (both PCV13 and PPSV23)
- Haemophilus influenzae type b (Hib)
- Meningococcal vaccines (both quadrivalent ACWY and type B)
- Annual influenza vaccination
- Post-Splenectomy Vaccination Schedule:
- Pneumococcal revaccination per guidelines
- Annual influenza vaccination
- Consideration of booster doses of meningococcal vaccines
- COVID-19 Vaccination:
- Generally recommended for ITP patients
- Monitoring of platelet counts after vaccination
- Individualized approach for patients with history of vaccine-associated ITP
- Pre-Splenectomy Vaccinations (ideally at least 2 weeks before surgery):
MMR Vaccination and ITP:
- Small risk of ITP following MMR vaccination (1 case per 40,000 vaccinations)
- Risk of ITP from natural measles infection much higher
- Individualized decision-making for children with history of ITP
- Generally not contraindicated in children with previous ITP
Preventive Screenings:
Regular Platelet Monitoring:
- Frequency determined by disease severity and treatment
- More frequent during treatment initiation or changes
- Can be reduced during periods of stability
Screening for Secondary Causes:
- Periodic reassessment for emerging associated conditions
- Age-appropriate cancer screenings, particularly for patients on long-term immunosuppression
- Monitoring for development of other autoimmune conditions
Bone Health Assessment:
- Bone density screening for patients on prolonged corticosteroid therapy
- Vitamin D level monitoring
- Fracture risk assessment
Cardiovascular Risk Assessment:
- Regular monitoring of blood pressure, lipids, and glucose, particularly for patients on corticosteroids
- Assessment of thrombotic risk, especially before TPO-RA therapy or splenectomy
Psychological Health Screening:
- Assessment for depression and anxiety
- Quality of life evaluation
- Functional status assessment
Patient Education:
- Recognition of bleeding warning signs
- When to seek emergency care
- Importance of medication adherence
- Informing all healthcare providers about ITP diagnosis
- Precautions regarding over-the-counter medications affecting platelet function
While primary prevention of ITP remains elusive, these preventive measures and lifestyle adaptations can significantly reduce complications and improve quality of life for patients living with this condition.
Global & Regional Statistics
Incidence and Prevalence Rates Globally
ITP is a relatively uncommon disorder with varying incidence and prevalence across different regions:
Global Incidence:
- The overall annual incidence of primary ITP is estimated at 1.9-6.4 cases per 100,000 person-years
- Children: 1.9-5.3 cases per 100,000 children per year
- Adults: 2-4 cases per 100,000 adults per year
- The incidence appears to have a bimodal age distribution with peaks in childhood (2-5 years) and later adulthood (>60 years)
Global Prevalence:
- Age-adjusted prevalence is approximately 9.5-12.1 cases per 100,000 persons
- Overall prevalence increases with age due to the chronic nature of adult ITP
- The prevalence in adults over 60 years of age may be as high as 20-25 per 100,000 persons
Regional Variations in Incidence:
North America:
- United States: Age-adjusted prevalence of 9.5 per 100,000 persons
- Canada: Annual incidence of approximately 2.9-3.9 per 100,000 persons
Europe:
- United Kingdom: Incidence of 1.6-2.9 per 100,000 person-years
- Denmark: Annual incidence of 2.68 per 100,000 persons
- France: Annual incidence of 2.9 cases per 100,000 person-years
- Northern Europe (overall): Annual incidence of 2.68 per 100,000 persons
Asia:
- Japan: Annual incidence of 2.0-3.1 per 100,000 persons
- Korea: Annual incidence of 3.2 per 100,000 person-years
- China: Limited nationwide data, but regional studies suggest incidence comparable to Western populations
Middle East:
- Kuwait: Higher reported incidence of childhood ITP at 125 cases per 1,000,000 children per year
- Israel: Annual incidence of approximately 2.6 per 100,000 persons
Africa:
- Limited data available
- Regional studies suggest potential higher rates in certain populations
- Significant underdiagnosis likely due to limited healthcare resources
Australia/Oceania:
- Australia: Annual incidence of 1.95-3.9 per 100,000 persons
- New Zealand: Similar rates to Australia
Factors Contributing to Regional Variations:
- Access to healthcare and diagnostic capabilities
- Genetic differences between populations
- Prevalence of associated infections (H. pylori, HIV, hepatitis C)
- Environmental exposures
- Reporting and surveillance systems
It’s important to note that substantial underreporting may occur in regions with limited healthcare access, and diagnostic criteria have evolved over time, potentially affecting incidence and prevalence estimates.
Mortality and Survival Rates
Mortality in ITP is primarily related to bleeding complications, particularly intracranial hemorrhage, though treatment-related complications and comorbidities also contribute:
Overall Mortality Rates:
- Children: <1% mortality rate, primarily from intracranial hemorrhage
- Adults: Approximately 5% overall mortality rate from ITP-related causes
- Elderly patients (>60 years) with severe thrombocytopenia: Significantly higher mortality rates
Mortality by Cause:
- Intracranial hemorrhage: Accounts for approximately 50% of ITP-related deaths
- Other major hemorrhage (gastrointestinal, pulmonary): 20-30% of ITP-related deaths
- Treatment complications: 10-15% of deaths
- Infections (particularly post-splenectomy): 10-15% of deaths
- Thrombotic events: Increasing recognition as a cause of mortality
Survival Rates:
- 5-year survival in chronic ITP: 90-95% in patients <40 years
- 5-year survival in chronic ITP: 70-80% in patients >60 years
- Predicted 5-year mortality rates from bleeding in patients with severe thrombocytopenia: 47.8% in patients >60 years vs. 2.2% in patients <40 years
Factors Affecting Survival:
- Age (strongest predictor of mortality)
- Platelet count (higher risk with counts <10,000/μL)
- Previous major bleeding episodes
- Comorbidities (especially cardiovascular disease)
- Response to treatment
- Time since diagnosis (highest risk in first year)
Long-term Outcomes:
- Children: >80% achieve spontaneous remission; excellent long-term survival
- Adults with persistent ITP: Approximately 50% achieve lasting remission with treatment
- Adults with chronic refractory ITP: Reduced life expectancy, primarily in elderly patients
Country-wise Comparison and Trends
ITP incidence, prevalence, and outcomes show distinct patterns across different countries and regions:
United States:
- Age-adjusted prevalence: 9.5-12.1 per 100,000 persons
- Female predominance in adults (1.7:1)
- Declining mortality rate over past decades with improved treatment
- Increasing use of TPO-RAs as second-line therapy
- Decreasing rates of splenectomy since introduction of TPO-RAs
- Healthcare expenditure for ITP: Approximately $28,000-$35,000 per patient annually
United Kingdom:
- Annual incidence: 1.6-2.9 per 100,000 person-years
- Strong registry data through UK Adult ITP Registry
- Guideline-driven care through British Society for Haematology
- Decreasing use of splenectomy (>50% reduction since 2005)
- Earlier use of TPO-RAs following NICE approval
Japan:
- Annual incidence similar to Western countries (2.0-3.1 per 100,000)
- Higher rates of H. pylori-associated ITP
- More favorable response to H. pylori eradication (>50% response)
- More cautious approach to splenectomy
- Strong research focus on immune mechanisms of ITP
China:
- Limited nationwide epidemiological data
- Higher rates of H. pylori infection and association with ITP
- Greater use of traditional Chinese medicine as adjunctive therapy
- Increasing access to novel therapies like TPO-RAs
- Significant urban-rural disparities in diagnosis and treatment
France:
- Annual incidence: 2.9 cases per 100,000 person-years
- Comprehensive national registry (CARMEN)
- Observed seasonal variation with winter peak and summer nadir
- Detailed data on secondary ITP (18% of adult cases)
- Persistence/chronicity in 36% of children vs. 67% of adults
Scandinavian Countries:
- Well-documented incidence through population-based registries
- Norway: Incidence of 53 per 1,000,000 in children <15 years
- Denmark: Increasing incidence with age, particularly >60 years
- Strong public health system facilitating comprehensive care
- Excellent long-term outcome data due to registry systems
Emerging Trends Across Countries:
Treatment Approaches:
- Global shift toward earlier use of TPO-RAs
- Declining rates of splenectomy worldwide
- Increasing use of rituximab as second-line therapy
- Growing focus on quality of life outcomes
Diagnostic Practices:
- More consistent application of standardized diagnostic criteria
- Less reliance on bone marrow examination
- Improved distinction between primary and secondary ITP
Healthcare Delivery:
- Development of specialized ITP centers of excellence
- Increased patient engagement through advocacy organizations
- Telehealth approaches for monitoring stable patients
Research Focus:
- International collaborative clinical trials
- Biomarker development for personalized therapy
- Focus on long-term outcomes and treatment-free remission
- Health-related quality of life research
While significant progress has been made in understanding the epidemiology of ITP globally, substantial gaps remain in data from developing regions, particularly in Africa, parts of Asia, and South America. International collaborative efforts are underway to improve global surveillance and standardize approaches to this relatively rare but significant disorder.
Recent Research & Future Prospects
Latest Advancements in Treatment and Research
The field of ITP has experienced significant advances in recent years, with several key developments transforming our understanding and management of the disease:
Thrombopoietin Receptor Agonists (TPO-RAs) Revolution:
- Treatment Paradigm Shift: TPO-RAs have moved from later lines of therapy to second-line and sometimes first-line treatment
- Expanded Indications: Originally approved for chronic ITP, now being used earlier in disease course (persistent ITP)
- Long-term Safety Data: Real-world evidence supporting safety with extended use (>10 years for romiplostim and eltrombopag)
- Treatment-Free Remission: Emerging evidence that 10-30% of patients can discontinue TPO-RAs and maintain stable platelet counts
- Newer Agents: Avatrombopag approval (2019) offering an additional oral option with fewer dietary restrictions than eltrombopag
Targeted Immunomodulation:
- Fostamatinib Approval: First-in-class Syk inhibitor approved for adult chronic ITP (2018)
- Rituximab Refinement: Lower-dose protocols showing similar efficacy with reduced side effects
- Bruton’s Tyrosine Kinase Inhibitors: Promising Phase II-III data for rilzabrutinib and others
- FcRn Antagonists: Novel mechanism targeting the neonatal Fc receptor to increase clearance of pathogenic antibodies
Improved Understanding of Pathophysiology:
- T-Cell Dysregulation: Recognition of cytotoxic CD8+ T-cell expansion as an antibody-independent mechanism
- Platelet Desialylation: Identification of Fc-independent mechanisms of platelet clearance
- Complement Activation: Evidence for complement involvement in platelet destruction
- Regulatory T-Cell Defects: Decreased number and function of regulatory T cells contributing to loss of immune tolerance
Biomarker Development:
- Predictive Biomarkers: Emerging markers to predict treatment response (e.g., immature platelet fraction for TPO-RA response)
- Prognostic Indicators: Identification of biomarkers associated with chronicity or treatment refractoriness
- Mechanistic Biomarkers: Markers differentiating antibody-mediated vs. T-cell-mediated disease
Advanced Diagnostics:
- Improved Antibody Testing: Development of more sensitive and specific tests for anti-platelet antibodies
- Glycoprotein-Specific Assays: Tests targeting specific platelet glycoproteins (GPIIb/IIIa, GPIb/IX)
- Genetic Profiling: Identification of genetic signatures associated with ITP subtypes
Treatment Strategies Refinement:
- Combination Approaches: Synergistic use of agents with complementary mechanisms of action
- Risk-Stratified Treatment: Tailoring therapy based on bleeding risk and patient characteristics
- Sequencing Studies: Optimal ordering of available therapies
- Early Intensive Therapy: Evaluation of aggressive initial treatment to induce long-term remission
Quality of Life Research:
- Validated Assessment Tools: Development of ITP-specific quality of life measures (ITP-PAQ)
- Patient-Reported Outcomes: Increased emphasis on fatigue, functional capacity, and emotional well-being
- Socioeconomic Impact Studies: Documentation of work productivity, healthcare utilization, and financial burden
Ongoing Studies and Future Medical Possibilities
Numerous exciting research initiatives are currently underway, with promising implications for future ITP management:
Active Clinical Trials:
Novel Therapeutic Agents:
- FcRn antagonists (rozanolixizumab, efgartigimod, nipocalimab)
- Next-generation BTK inhibitors (tolebrutinib, evobrutinib)
- Complement inhibitors (sutimlimab targeting C1s, pegcetacoplan targeting C3)
- Plasma cell targeting agents (daratumumab, proteasome inhibitors)
Treatment Strategy Studies:
- Early combined immunosuppression vs. sequential therapy
- Predictors of TPO-RA treatment-free remission
- Intermittent TPO-RA dosing strategies
- Risk-adapted treatment algorithms
Pediatric-Focused Research:
- TPO-RA safety and efficacy in children
- Predictors of chronicity in childhood ITP
- Minimal treatment approaches for low-risk children
- Long-term neurodevelopmental outcomes after childhood ITP
Special Populations:
- Pregnancy registry studies
- Management strategies for elderly patients
- Secondary ITP treatment approaches
- COVID-19 vaccine-associated ITP
Future Therapeutic Approaches:
Cell-Based Therapies:
- Regulatory T-Cell (Treg) Therapy: Expansion or infusion of regulatory T cells to restore immune tolerance
- Chimeric Antigen Receptor (CAR) T-Cell Therapy: Targeting autoreactive B cells
- Mesenchymal Stem Cell Therapy: Immunomodulatory and regenerative effects
- Tolerogenic Dendritic Cell Therapy: Induction of immune tolerance
Gene Therapy and Editing:
- Correction of genetic susceptibility factors
- Regulation of immune activation pathways
- Enhancement of thrombopoiesis
- RNA interference targeting pathogenic immune pathways
Precision Medicine Approaches:
- Disease Mechanism Classification: Treatment selection based on predominant pathophysiology (antibody-mediated vs. T-cell-mediated)
- Genetic Profile-Based Therapy: Treatment guided by genetic susceptibility factors
- Response Prediction Models: Algorithms to predict optimal treatment for individual patients
- Biomarker-Guided Treatment Decisions: Use of laboratory markers to personalize therapy
Novel Drug Delivery Systems:
- Nanoparticle-based targeted delivery
- Long-acting formulations reducing dosing frequency
- Oral alternatives to injectable therapies
- Combination drug platforms
Future Diagnostic Advances:
Point-of-Care Testing:
- Rapid platelet function testing
- Home monitoring of platelet counts
- Portable coagulation assessment tools
Advanced Imaging:
- Novel tracers for assessing platelet production and destruction
- Molecular imaging of immune activation
- Non-invasive assessment of bleeding risk
Artificial Intelligence Applications:
- Pattern recognition in clinical presentations
- Prediction of disease trajectory
- Optimal treatment selection
- Early identification of complications
Potential Cures or Innovative Therapies Under Development
While ITP has historically been considered a manageable but not curable condition, several approaches under investigation hold promise for potential curative strategies:
Disease-Modifying Approaches:
Immune Tolerance Induction:
- Antigen-Specific Immunotherapy: Targeting immune responses to specific platelet antigens
- Co-stimulation Blockade: Prevention of aberrant T-cell activation
- Regulatory T-Cell Expansion: Restoration of immune tolerance
- Status: Early-phase clinical trials and preclinical development
Early Intervention Protocols:
- Combination Immunomodulation: Synchronized targeting of multiple immune pathways
- Intensive Initial Therapy: Aiming to “reset” the immune system
- Targeted Plasma Cell Depletion: Elimination of autoantibody-producing cells before long-lived plasma cell establishment
- Status: Phase II-III trials evaluating early combination approaches
Microbiome Modulation:
- Gut Microbiome Restoration: Addressing potential microbial triggers of autoimmunity
- Microbiota-Derived Metabolites: Novel immunomodulatory compounds
- Fecal Microbiota Transplantation: Being explored in various autoimmune conditions
- Status: Primarily preclinical with early translational studies
Potential Curative Approaches:
Hematopoietic Stem Cell Transplantation:
- Autologous Transplantation with Immunoablation: Elimination and “resetting” of the immune system
- Allogeneic Transplantation: Replacement with donor immune system
- Status: Limited to severe, refractory cases due to risk; case reports of long-term remission
Targeted Gene Editing:
- CRISPR-Cas9 Applications: Correction of genetic susceptibility
- B-Cell Receptor Editing: Elimination of autoreactive B-cell clones
- Status: Early preclinical research
Next-Generation Cellular Therapies:
- Engineered Regulatory T Cells: Enhanced suppressive capacity
- Chimeric Antigen Receptor Regulatory T Cells (CAR-Tregs): Targeted immunosuppression
- Platelet-Producing Organoids: Ex vivo platelet generation systems
- Status: Preclinical development with early clinical applications in other autoimmune conditions
Barriers to Curative Approaches:
Disease Heterogeneity:
- Multiple pathogenic mechanisms requiring tailored approaches
- Lack of definitive diagnostic biomarkers for disease subtypes
- Challenge of predicting which patients might benefit from intensive therapies
Established Immune Memory:
- Long-lived plasma cells resistant to many therapies
- Persistent autoreactive memory B and T cells
- Difficulty in selectively targeting pathogenic cells while preserving protective immunity
Risk-Benefit Considerations:
- Current treatments effectively manage most cases with acceptable safety profiles
- Higher-risk curative approaches must demonstrate significant advantage
- Ethical considerations in exposing patients to experimental therapies
Regulatory and Economic Challenges:
- Complexity of clinical trial design for rare disorders
- High development costs for personalized approaches
- Reimbursement challenges for complex cellular therapies
Despite these challenges, the convergence of advances in immunology, cell therapy, and precision medicine offers unprecedented opportunities for transformative therapies. The goal of achieving long-term treatment-free remission, if not outright cure, appears increasingly feasible for at least a subset of ITP patients in the coming decade.
Interesting Facts & Lesser-Known Insights
Uncommon Knowledge About ITP
Beyond the standard medical understanding, several fascinating and lesser-known aspects of ITP provide deeper insight into this complex disorder:
Historical Curiosities:
The “Harrington-Hollingsworth Experiment” in 1950, where Dr. William Harrington injected himself with blood from an ITP patient and developed severe thrombocytopenia, remains one of the most dramatic self-experimentation episodes in modern medical history. Harrington’s interest in ITP was sparked by caring for a 17-year-old girl who died from bleeding after splenectomy.
Before the disease was understood, various folk remedies for purpura included applications of leeches, consumption of liver extracts, and exposure to certain mineral springs, some of which persisted into the early 20th century.
The term “purpura” derives from the Latin word for “purple,” which itself comes from the Greek “porphyra,” referring to a sea snail that produced a valuable purple dye in ancient times.
Biological Peculiarities:
ITP is one of few autoimmune conditions where the primary target (platelets) has a very short lifespan (7-10 days) even under normal conditions. This rapid turnover contributes to the often dramatic response to therapies.
Despite having severely reduced platelet counts, many ITP patients experience little or no bleeding. This “asymptomatic thrombocytopenia” phenomenon is not fully understood but suggests compensatory mechanisms that enhance the function of remaining platelets.
“Rebound thrombocytosis” can occur after successful ITP treatment, with platelet counts temporarily exceeding normal limits, creating a theoretical risk of thrombosis during recovery.
Some research suggests circadian rhythms affect platelet counts, with counts potentially varying by 5-10% throughout the day. This may have implications for timing of blood draws and interpretation of results.
Treatment Curiosities:
Before the development of modern therapies, unusual treatments for ITP included arsenic compounds, irradiation of the spleen, and whole blood transfusions from patients with thrombocytosis.
Some patients report unexpected remissions following acute infections or fevers, suggesting that alterations in immune regulation during infection may sometimes beneficially reset immune tolerance.
The anti-malarial drug hydroxychloroquine, better known for treating lupus and rheumatoid arthritis, shows efficacy in some ITP patients, particularly those with antinuclear antibodies or other autoimmune features.
Helicobacter pylori eradication therapy leads to platelet count improvement in 50-60% of ITP patients in Japan but only 10-15% in North America, suggesting genetic or environmental differences in disease mechanism.
Paradoxical Features:
Despite being a bleeding disorder, ITP patients have an approximately 1.5-2 times higher risk of thrombosis (blood clots) compared to the general population, creating a complex balance in management decisions.
The spleen, while a major site of platelet destruction in ITP, is typically not enlarged. In fact, splenomegaly should prompt consideration of alternative diagnoses.
TPO-RA therapy can lead to treatment-free remissions in 10-30% of chronic ITP patients, suggesting these agents may have immunomodulatory effects beyond simply increasing platelet production.
Myths and Misconceptions vs. Medical Facts
Several misconceptions persist about ITP among both the general public and sometimes within healthcare communities:
Myth: ITP is primarily a childhood disease. Fact: While ITP is common in children, the incidence in adults is similar. The adult form is more likely to be chronic and less likely to resolve spontaneously.
Myth: All patients with ITP have obvious bleeding symptoms. Fact: Many patients, even with very low platelet counts (<10,000/μL), may have minimal or no bleeding manifestations. The correlation between platelet count and bleeding is imperfect.
Myth: ITP is caused by vitamin K deficiency. Fact: Vitamin K deficiency affects clotting factors, not platelets. ITP is an immune-mediated disorder unrelated to vitamin deficiencies.
Myth: ITP is contagious. Fact: ITP is an autoimmune disorder that cannot be transmitted from person to person. While certain infections may trigger ITP, the condition itself is not contagious.
Myth: Platelet transfusions are a standard treatment for ITP. Fact: Platelet transfusions are generally avoided in ITP except for life-threatening bleeding, as transfused platelets are rapidly destroyed by the same autoimmune process.
Myth: ITP requires immediate emergency treatment whenever diagnosed. Fact: Many patients with mild to moderate thrombocytopenia without significant bleeding can be safely observed without immediate intervention.
Myth: All ITP patients eventually need splenectomy. Fact: With modern therapies, particularly TPO-RAs, the need for splenectomy has dramatically decreased. Many patients achieve good control without surgical intervention.
Myth: A low platelet count always means ITP. Fact: Many conditions can cause thrombocytopenia, including medications, infections, other autoimmune diseases, and bone marrow disorders. ITP is a diagnosis of exclusion.
Myth: ITP always requires lifelong treatment. Fact: Many children (80-90%) and some adults (approximately 20-30%) experience spontaneous remission without the need for ongoing therapy.
Myth: Patients with ITP should strictly avoid all physical activity. Fact: Activity restrictions should be individualized based on platelet count, bleeding history, and the nature of the activity. Many patients can maintain normal or near-normal activity with appropriate precautions.
Impact on Specific Populations or Professions
ITP affects various populations differently and can have unique implications for certain professions or life circumstances:
Professional Impacts:
High-Risk Occupations:
- Military Personnel: May face medical discharge or duty restrictions due to bleeding risk and medication requirements
- Professional Athletes: Contact sports may be contraindicated with severe thrombocytopenia
- Construction and Heavy Machine Operators: Heightened risk due to potential trauma
- Surgeons and Dentists: Fine motor procedures may be affected, particularly during disease flares
- Commercial Pilots: Licensing restrictions related to certain ITP medications and risk of sudden incapacitation
Special Management Considerations by Population:
Pregnant Women:
- ITP complicates 1-10 pregnancies per 10,000
- Maternal platelet autoantibodies can cross the placenta and affect the fetus
- 10-20% of neonates born to mothers with ITP develop transient thrombocytopenia
- Management challenges include:
- Limited treatment options (many drugs contraindicated)
- Need to maintain platelets >30,000/μL during pregnancy and >50,000/μL for delivery
- Interdisciplinary care involving hematology, obstetrics, and neonatology
- Potential for neonatal thrombocytopenia requiring monitoring
Elderly Patients:
- Higher risk of bleeding at equivalent platelet counts compared to younger patients
- Greater concern for treatment-related complications, particularly with corticosteroids
- Often have comorbidities that complicate management (cardiovascular disease, diabetes)
- Higher risk of drug interactions due to polypharmacy
- Potential cognitive and functional impacts affecting treatment adherence
Adolescents:
- Challenging transition period from pediatric to adult care
- Body image concerns with steroid therapy
- Impact on education and social development
- Issues with treatment adherence and risk-taking behavior
- Need for specialized transition programs
Ethnicity-Specific Considerations:
- Higher prevalence of H. pylori in Asian populations and greater response to eradication therapy
- Potential differences in drug metabolism affecting treatment response and side effects
- Cultural beliefs affecting healthcare-seeking behavior and treatment adherence
- Genetic factors influencing disease manifestation and treatment response
Socioeconomic Impacts:
Employment Challenges:
- 49% of ITP patients report reducing or considering reducing working hours
- 11% forced to stop working entirely due to ITP
- Higher rates (19%) of employment cessation in those with severe symptom burden
- Difficulties with workplace accommodations and disability benefits
Financial Burden:
- High medication costs, particularly for novel agents (TPO-RAs can cost >$100,000 annually)
- Frequent medical visits and laboratory monitoring
- Potential loss of income and career advancement
- Insurance coverage challenges for off-label treatments
Healthcare Disparities:
- Access to specialized care varies significantly by region
- Medication availability differs across healthcare systems
- Rural patients may face challenges accessing ITP specialists
- Disparities in outcomes based on socioeconomic status and healthcare access
Understanding these nuanced aspects of ITP contributes to more compassionate and comprehensive care for affected individuals, recognizing that the impact extends far beyond the medical manifestations to affect many dimensions of life and functioning.
References
American Society of Hematology. (2019). ASH Clinical Practice Guidelines on Immune Thrombocytopenia. Blood Advances, 3(23), 3829-3866.
Cines, D. B., & Bussel, J. B. (2005). How I treat idiopathic thrombocytopenic purpura (ITP). Blood, 106(7), 2244-2251.
Cooper, N., & Ghanima, W. (2019). Immune Thrombocytopenia. New England Journal of Medicine, 381(10), 945-955.
Frederiksen, H., & Schmidt, K. (1999). The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood, 94(3), 909-913.
Harrington, W. J., Minnich, V., Hollingsworth, J. W., & Moore, C. V. (1951). Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. The Journal of Laboratory and Clinical Medicine, 38(1), 1-10.
International Working Group. (2009). Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood, 113(11), 2386-2393.
Kuter, D. J. (2019). The treatment of immune thrombocytopenia (ITP)—focus on thrombopoietin receptor agonists. Annals of Blood, 4, 10.
Moulis, G., Palmaro, A., Montastruc, J. L., Godeau, B., Lapeyre-Mestre, M., & Sailler, L. (2014). Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood, 124(22), 3308-3315.
Neunert, C., Terrell, D. R., Arnold, D. M., Buchanan, G., Cines, D. B., Cooper, N., … & Ghanima, W. (2019). American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Advances, 3(23), 3829-3866.
Provan, D., Arnold, D. M., Bussel, J. B., Chong, B. H., Cooper, N., Gernsheimer, T., … & Kuter, D. J. (2019). Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Advances, 3(22), 3780-3817.
Rodeghiero, F., Stasi, R., Gernsheimer, T., Michel, M., Provan, D., Arnold, D. M., … & Cines, D. B. (2009). Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood, 113(11), 2386-2393.
Segal, J. B., & Powe, N. R. (2006). Prevalence of immune thrombocytopenia: analyses of administrative data. Journal of Thrombosis and Haemostasis, 4(11), 2377-2383.
Stasi, R. (2011). ITP: a historical perspective. British Journal of Haematology, 153(4), 437-450.
Terrell, D. R., Beebe, L. A., Vesely, S. K., Neas, B. R., Segal, J. B., & George, J. N. (2010). The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. American Journal of Hematology, 85(3), 174-180.
Zeller, B., Helgestad, J., Hellebostad, M., Kolmannskog, S., Nystad, T., Stensvold, K., & Wesenberg, F. (2000). Immune thrombocytopenic purpura in childhood in Norway: a prospective, population-based registration. Pediatric Hematology and Oncology, 17(7), 551-558.