
⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
Comprehensive Report on Polycystic Kidney Disease
1. Overview
What is Polycystic Kidney Disease?
Polycystic Kidney Disease (PKD) is a genetic disorder characterized by the development of numerous fluid-filled cysts in the kidneys. These cysts vary in size from microscopic to several centimeters in diameter and gradually replace normal kidney tissue, resulting in progressive kidney enlargement and declining kidney function. PKD is not a single disease but rather a collection of inherited disorders with similar kidney manifestations.
The two primary forms of PKD are:
Autosomal Dominant Polycystic Kidney Disease (ADPKD): The most common form, typically manifesting in adulthood.
Autosomal Recessive Polycystic Kidney Disease (ARPKD): A rarer, more severe form that often presents in infancy or early childhood.
Affected Body Parts/Organs:
While the kidneys are the primary affected organs, PKD is a systemic disorder that can impact multiple organ systems:
- Kidneys: Progressive cyst development, enlargement, and eventual function impairment
- Liver: Development of bile duct cysts (affecting up to 90% of ADPKD patients)
- Pancreas: Cystic lesions (less common)
- Cardiovascular system: Vascular abnormalities, including intracranial aneurysms and heart valve defects
- Gastrointestinal tract: Diverticulosis and abdominal wall hernias
- Reproductive organs: Seminal vesicle cysts in men
Prevalence and Significance:
PKD represents one of the most common life-threatening genetic diseases:
- ADPKD affects approximately 1 in 400 to 1,000 people worldwide, making it the most common inherited kidney disease
- ADPKD accounts for 5-10% of all cases of end-stage renal disease (ESRD) requiring dialysis or transplantation
- ARPKD is much rarer, affecting approximately 1 in 20,000 live births
- Global prevalence estimates suggest 12.5 million people are affected by PKD
- The disease has significant economic impact, with annual healthcare costs for ADPKD patients estimated at $50,000-$100,000 per patient in advanced stages
- PKD is a leading genetic cause of kidney transplantation
The progressive nature of PKD, its systemic manifestations, and its impact on quality of life make it a significant public health concern and an important focus for research into genetic therapies and targeted treatments.
2. History & Discoveries
The medical understanding of Polycystic Kidney Disease has evolved dramatically over the past two centuries, from anatomical observations to sophisticated molecular genetic insights.
Early Identification and Description:
1820s: Early descriptions of polycystic kidneys were documented in medical literature, initially considered rare curiosities rather than a specific disease entity.
1888: Felix Lejars published the first comprehensive report distinguishing adult polycystic kidney disease from other renal cystic disorders.
1899: William Osler recognized the hereditary nature of the condition, describing its familial pattern.
1900-1910: Multiple pathologists documented the characteristic gross and microscopic features of PKD, establishing it as a distinct clinicopathological entity.
Recognition as a Genetic Disorder:
1950s: Dalgaard conducted pioneering epidemiological studies confirming the autosomal dominant inheritance pattern of ADPKD.
1957: Becker and colleagues definitively established the difference between the adult (dominant) and infantile (recessive) forms of PKD.
1970s: Improved imaging techniques, particularly ultrasound, revolutionized the non-invasive diagnosis of PKD, enabling family studies and earlier detection.
Genetic Breakthroughs:
1985: The first genetic linkage for ADPKD was established on chromosome 16, suggesting a specific genetic locus (later identified as PKD1).
1994: The PKD1 gene was identified and sequenced by the European Polycystic Kidney Disease Consortium, representing a landmark achievement in understanding the molecular basis of ADPKD.
1996: The second gene for ADPKD, PKD2, was discovered, explaining cases not linked to PKD1 mutations.
2002: PKHD1, the gene responsible for ARPKD, was identified, completing the genetic picture of the major forms of PKD.
2007-2010: Multiple studies elucidated the roles of polycystin-1 and polycystin-2 (the protein products of PKD1 and PKD2) in cellular signaling and ciliary function.
Treatment Milestones:
1960s-1970s: Introduction of hemodialysis and kidney transplantation offered effective management for end-stage kidney disease in PKD patients.
2000s: Understanding of the pathophysiology led to the identification of vasopressin’s role in cyst growth, setting the stage for targeted therapies.
2018: FDA approval of tolvaptan (Jynarque), the first medication specifically indicated to slow kidney function decline in ADPKD, marking a new era in PKD treatment.
Evolution of Medical Understanding:
The conceptualization of PKD has transformed through several paradigms:
Anatomical Period (1800s-1950s): Focus on kidney morphology and clinical manifestations.
Genetic Era (1950s-1990s): Establishment of inheritance patterns and family screening protocols.
Molecular Age (1990s-2000s): Identification of causative genes and their protein products.
Pathophysiology Revolution (2000s-present): Understanding of cellular mechanisms, ciliary dysfunction, and signaling pathways.
Therapeutic Era (2010s-present): Development of targeted treatments based on molecular understanding.
The history of PKD research exemplifies how advances in genetics, molecular biology, and imaging technologies have transformed our understanding of a disease from purely descriptive to mechanistic, ultimately leading to therapeutic interventions targeting the underlying pathophysiology.
3. Symptoms
Polycystic Kidney Disease spans a spectrum of clinical manifestations that typically progress over time, with significant variation in both severity and rate of progression among affected individuals.
Early Symptoms:
Many individuals with ADPKD remain asymptomatic for decades, with the disease often being discovered incidentally during imaging studies performed for unrelated reasons. Early symptoms, when present, include:
- Hypertension: Often the earliest manifestation, occurring in 60-70% of patients before significant kidney function decline, typically appearing in the 20s or 30s
- Pain: Dull, aching flank or abdominal pain, usually related to kidney enlargement or cyst hemorrhage
- Urinary Tract Infections (UTIs): More common than in the general population
- Hematuria: Microscopic or gross (visible) blood in urine, often following physical exertion or cyst rupture
- Kidney Stones: Occur in approximately 20% of patients
- Subtle Urinary Concentration Defects: Increased urination frequency or nocturia (nighttime urination)
For ARPKD, which typically manifests in infancy or childhood:
- Enlarged kidneys: Often detectable prenatally by ultrasound
- Pulmonary hypoplasia: Breathing difficulties in severe neonatal cases
- Hypertension: Present in up to 80% of affected children
- Growth retardation: Related to kidney dysfunction
Advanced-Stage Symptoms:
As PKD progresses, symptoms become more pronounced and complications more frequent:
Signs of Declining Kidney Function:
- Fatigue and weakness
- Poor appetite
- Swelling in legs, ankles, and feet (edema)
- Decreased urine output or changes in urination patterns
- Difficulty concentrating and cognitive changes
Increased Pain and Discomfort:
- Chronic abdominal distension
- More frequent and severe pain episodes
- Physical limitations due to enlarged kidneys
Manifestations of End-Stage Renal Disease (ESRD):
- Uremic symptoms (nausea, vomiting, metallic taste)
- Pruritus (itching)
- Restless legs syndrome
- Sleep disturbances
- Pericarditis (inflammation of the heart lining)
- Uremic frost (rare)
Common vs. Rare Symptoms:
Common Symptoms (>30% of patients):
- Hypertension (70-80%)
- Flank pain (60%)
- Hematuria (35-50%)
- Urinary tract infections (30-50%)
- Abdominal fullness or distension (40%)
Less Common Symptoms (10-30%):
- Kidney stones (20%)
- Liver cyst complications (25% of those with liver cysts)
- Back pain related to kidney enlargement (20%)
- Gout (15-20%)
Rare Symptoms (<10%):
- Cerebral aneurysm rupture (5-10%)
- Diverticular disease complications (5%)
- Mitral valve prolapse symptoms (5%)
- Pancreatic cyst-related symptoms (1-5%)
- Hernias (5-10%)
- Seminal vesicle cysts causing male infertility (rare)
Symptom Progression Over Time:
The typical trajectory of ADPKD follows these general phases, though with considerable individual variation:
Preclinical Phase (Birth to age 15-20):
- Microscopic cyst formation
- No detectable symptoms
- Normal kidney function
Early Clinical Phase (Age 20-40):
- Detectable kidney enlargement
- Hypertension emergence
- Episodes of pain or hematuria
- Preserved kidney function
Progressive Phase (Age 40-60):
- Significant kidney enlargement (often >1000mL volume per kidney)
- Declining kidney function
- Increasing hypertension severity
- More frequent pain episodes
- Growing impact on quality of life
End-Stage Phase (Typically age 55-75):
- Kidney failure requiring renal replacement therapy
- Systemic complications
- Maximum kidney enlargement
For ARPKD, the progression pattern depends on the severity at presentation:
- Neonatal severe form may lead to early mortality
- Infantile forms show progressive kidney and liver dysfunction
- Juvenile forms may have slower progression with both kidney and liver manifestations
The heterogeneity in symptom presentation and progression underscores the importance of individualized monitoring and management strategies for PKD patients, with regular assessment of kidney function, blood pressure, and development of complications.
4. Causes
Polycystic Kidney Disease is primarily a genetic disorder, with specific mutations driving the disease process, though various factors may influence disease expression and progression.
Genetic Basis:
1. Autosomal Dominant Polycystic Kidney Disease (ADPKD):
Results from mutations in one of two primary genes:
- PKD1 gene (chromosome 16): Accounts for approximately 78% of cases
- PKD2 gene (chromosome 4): Accounts for approximately 15% of cases
- GANAB, DNAJB11, and other genes: Account for the remaining cases
Inheritance pattern:
- Only one copy of the mutated gene is needed to cause the disease
- Each child of an affected parent has a 50% chance of inheriting the mutation
- Approximately 10% of cases result from new (de novo) mutations with no family history
Genetic mechanisms:
- PKD1 encodes polycystin-1, a large transmembrane protein
- PKD2 encodes polycystin-2, a calcium channel protein
- These proteins interact at the primary cilium of kidney cells
- Mutation leads to disrupted calcium signaling and increased cell proliferation
2. Autosomal Recessive Polycystic Kidney Disease (ARPKD):
Caused by mutations in the PKHD1 gene on chromosome 6
Inheritance pattern:
- Both parents must be carriers of the mutation
- Each child has a 25% chance of developing the disease
- 50% chance of being an asymptomatic carrier
- 25% chance of not receiving the mutation
Genetic mechanisms:
- PKHD1 encodes fibrocystin/polyductin, a ciliary protein
- Dysfunction leads to abnormal tubular development and cyst formation
Biological Mechanisms of Cyst Formation:
The pathogenesis of PKD involves multiple cellular processes:
Ciliary Dysfunction: Primary cilia on kidney epithelial cells serve as sensory organelles. Mutations in PKD genes impair ciliary function, disrupting key signaling pathways.
Disrupted Calcium Signaling: Polycystins regulate calcium entry into cells. Dysfunction leads to reduced intracellular calcium, affecting multiple downstream pathways.
Increased Cell Proliferation: Mutated cells show increased proliferation rates and resistance to apoptosis (programmed cell death).
Fluid Secretion: Abnormal chloride and fluid transport into the developing cysts causes progressive enlargement.
Extracellular Matrix Abnormalities: Changes in basement membrane composition contribute to cyst growth.
Activation of mTOR and MAPK Pathways: These cellular signaling pathways become dysregulated, promoting cyst growth.
cAMP Signaling: Elevated levels of cyclic AMP promote both fluid secretion and cell proliferation.
Two-Hit Hypothesis and Mosaicism:
For ADPKD, the “two-hit hypothesis” helps explain the focal nature of cyst development:
- Individuals inherit one mutated copy of PKD1 or PKD2
- Random somatic (acquired) mutations in the normal copy in individual cells serve as the “second hit”
- Only cells experiencing both events develop cysts
- This explains why only 1-2% of nephrons develop cysts despite all cells carrying the germline mutation
Environmental and Modifying Factors:
While PKD is primarily genetic, several factors may influence disease progression:
Vasopressin Activity: Higher levels of this hormone accelerate cyst growth by increasing cAMP production.
Dietary Factors:
- High sodium intake may worsen hypertension and disease progression
- Protein intake may influence progression rate
- Caffeine potentially stimulates cyst growth through cAMP pathways
Hydration Status: Some evidence suggests that increased water intake may slow progression by suppressing vasopressin.
Gender: Male sex is associated with more rapid progression in many studies.
Kidney Injury: Ischemic or toxic injuries may accelerate cyst formation and growth.
Genetic Modifiers: Additional genes may influence disease severity and progression rate.
PKD Gene Type and Position: PKD1 mutations generally cause more severe disease than PKD2, and truncating mutations typically cause more severe disease than missense mutations.
The complex interplay between genetic factors and environmental influences explains the significant variability in disease presentation and progression, even among members of the same family with identical PKD mutations. This understanding has led to targeted therapeutic approaches aimed at modifying the signaling pathways involved in cyst formation and growth.
5. Risk Factors
The risk profile for Polycystic Kidney Disease is dominated by genetic factors, but several elements influence who develops the disease, when symptoms appear, and how quickly it progresses.
Primary Risk Factors:
1. Family History and Genetic Factors:
- ADPKD: Having a parent with ADPKD confers a 50% risk of inheriting the condition
- ARPKD: Both parents must be carriers, resulting in a 25% risk for each child
- Approximately 10% of ADPKD cases occur without family history due to new mutations
- Genetic background and modifier genes may influence disease severity
2. Gene Type and Mutation Location:
- PKD1 mutations generally cause more severe disease than PKD2:
- PKD1: Average age of ESRD around 54-56 years
- PKD2: Average age of ESRD around 74 years
- Truncating mutations (those that result in incomplete proteins) typically cause more severe disease than missense mutations
- Position of mutation within the gene affects severity
3. Age:
- ADPKD manifestations increase with age:
- <30 years: 30% have detectable cysts on imaging
- 30-59 years: 75% have detectable cysts
60 years: Nearly 100% penetrance in mutation carriers
- ARPKD severity often correlates inversely with age at diagnosis:
- Neonatal presentation typically indicates severe disease
- Later childhood diagnosis often indicates milder course
4. Gender Influences:
- Males typically experience faster progression to ESRD (approximately 5-10 years earlier than females)
- Females have higher prevalence of liver cysts and more extensive hepatic involvement
- Pregnancy may accelerate kidney growth in women with ADPKD
Factors Affecting Disease Progression:
1. Environmental Factors:
- Hydration Status: Chronic dehydration increases vasopressin levels, potentially accelerating cyst growth
- Dietary Patterns:
- High sodium intake worsens hypertension and may accelerate progression
- High protein intake may increase hyperfiltration and accelerate decline
- Caffeine consumption potentially stimulates cyst growth
- Smoking: Associated with faster progression and earlier onset of ESRD
- Obesity: BMI >30 correlates with faster GFR decline in some studies
2. Pre-existing Medical Conditions:
- Hypertension: Both a consequence and accelerator of PKD
- Urinary Tract Infections: Repeated infections may damage kidney tissue
- Kidney Stones: Can cause obstruction and accelerate function decline
- Diabetes: Comorbid diabetic nephropathy can hasten progression
- Cardiovascular Disease: Adds to overall risk profile and mortality
3. Genetic Modifiers:
- HLA Type: Certain HLA haplotypes associated with more aggressive disease
- Angiotensin-Converting Enzyme (ACE) Gene Polymorphisms: May influence progression rate
- Variants in Metabolic Pathway Genes: Emerging evidence for their modifying effect
4. Occupational and Lifestyle Considerations:
- Physical Trauma Risk: Contact sports or high-impact activities may increase risk of cyst rupture and bleeding
- Occupations Requiring Specific Medical Standards: May be affected by PKD diagnosis (pilots, military personnel)
- Limited Access to Healthcare: Delayed diagnosis and treatment accelerate progression
5. Pregnancy-Related Factors:
- Pregnancy complications occur in 35% of women with ADPKD
- Hypertensive disorders of pregnancy more common (15-30%)
- Each pregnancy associated with slight acceleration in kidney growth
Risk Assessment and Prognostic Models:
Several tools have been developed to assess risk and predict progression:
Mayo Imaging Classification: Based on height-adjusted total kidney volume (htTKV) and age
- Class 1A: Slowest progression
- Class 1E: Fastest progression
PROPKD Score: Incorporates:
- Gender
- Hypertension onset before age 35
- Urological complications before age 35
- Genotype (PKD1 truncating vs. PKD1 non-truncating vs. PKD2)
Kidney Failure Risk Equation: Modified for PKD to predict progression to ESRD
Understanding risk factors is crucial for patient counseling, treatment planning, and clinical trial design. The identification of modifiable risk factors has led to lifestyle recommendations that may complement pharmacological approaches to slow disease progression.
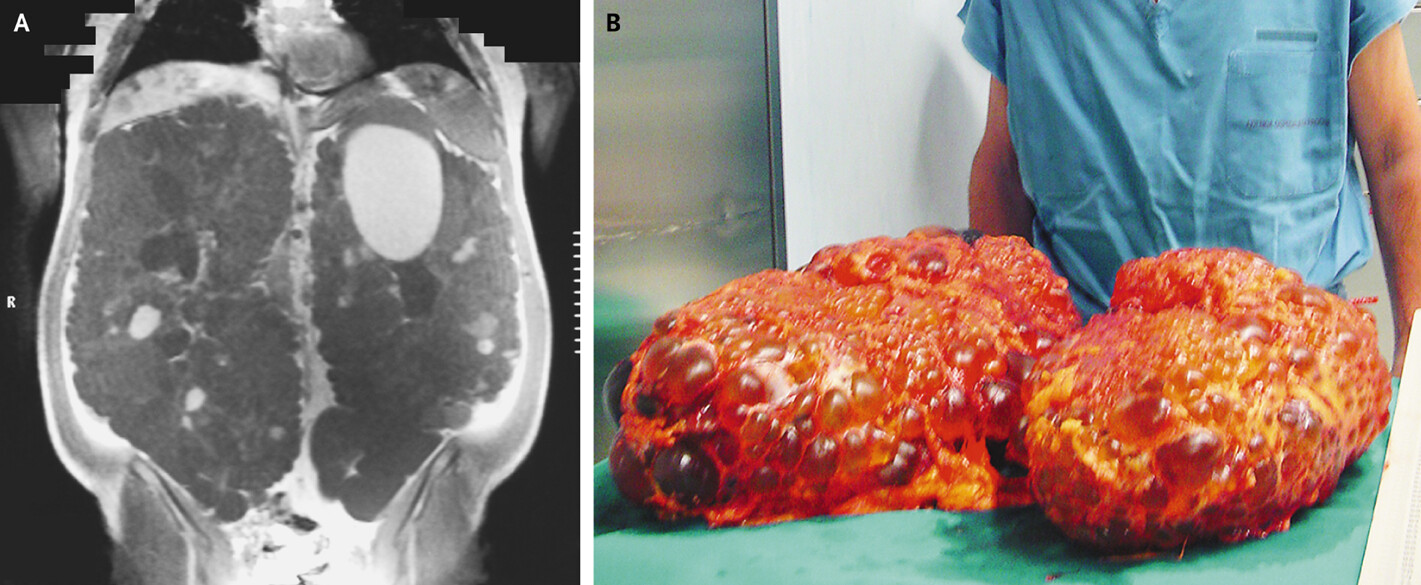
6. Complications
Polycystic Kidney Disease can affect multiple organ systems beyond the kidneys, leading to diverse complications that significantly impact morbidity, mortality, and quality of life.
Kidney-Related Complications:
1. Progressive Kidney Failure:
- 45-70% of ADPKD patients develop end-stage renal disease (ESRD) by age 65
- Median age of ESRD onset: 54-56 years for PKD1, 74 years for PKD2
- Requires renal replacement therapy (dialysis or transplantation)
- Associated with significant mortality risk and reduced quality of life
2. Hypertension:
- Affects 60-80% of patients before significant kidney function decline
- Often develops in early adulthood (20s-30s)
- More severe and earlier onset than in general population
- Contributes to cardiovascular morbidity and accelerates kidney disease progression
- Results from renin-angiotensin-aldosterone system activation and vascular compression
3. Urological Complications:
- Kidney Stones: Occur in 20-35% of patients
- Caused by urinary stasis, metabolic abnormalities, and anatomical changes
- May cause obstruction and accelerate function decline
- Urinary Tract Infections: 30-50% lifetime risk
- Can extend to cyst infection (difficult to treat)
- May trigger cyst hemorrhage
- Hematuria: Affects up to 50% of patients
- Gross hematuria episodes in 30-40% of patients
- Usually self-limiting but can be severe
4. Pain Syndromes:
- Chronic pain affects 60% of patients
- Acute episodes related to cyst rupture, infection, or stone passage
- Can become debilitating and affect employment and quality of life
- Pain management is a major clinical challenge
Extrarenal Complications:
1. Hepatic Manifestations:
- Liver cysts present in 75-90% of patients by age 60
- More common and extensive in women, especially with multiple pregnancies
- Can cause:
- Massive hepatomegaly
- Bile duct compression
- Portal hypertension (rare)
- Hepatic cyst infection
- Symptomatic in approximately 20% of patients
- Rarely progresses to liver failure
2. Cardiovascular Complications:
Intracranial Aneurysms: Present in 8-12% of ADPKD patients
- Family clustering observed
- Rupture risk approximately 0.1% per year
- Rupture carries 35-55% mortality risk
Heart Valve Abnormalities:
- Mitral valve prolapse in 25-30% of patients
- Aortic insufficiency in 5-10%
Aortic Root Dilation and Aneurysms:
- Related to underlying connective tissue abnormalities
- Present in 5-10% of patients
Coronary Artery Disease:
- Accelerated by hypertension and kidney disease
- Major cause of mortality
3. Gastrointestinal Complications:
- Diverticular Disease: Higher prevalence and complication rates
- Abdominal Wall Hernias: 15-20% of patients
- Pancreatic Cysts: 5-10% of patients, rarely symptomatic
4. Reproductive System Manifestations:
- Seminal Vesicle Cysts: Can affect male fertility
- Pregnancy Complications:
- Increased hypertensive disorders (20-30%)
- Higher risk of preeclampsia
- More frequent urinary tract infections
Psychological and Quality of Life Impact:
- Depression and anxiety: 25-30% prevalence
- Body image concerns due to abdominal distension
- Sleep disturbances (pain, nocturia)
- Sexual dysfunction
- Employment limitations and disability
- Financial burden of chronic disease management
Mortality and Long-term Outcomes:
- Standardized mortality ratio: 1.6-3.2 (higher in younger patients)
- Leading causes of death:
- Cardiovascular disease (36%)
- Infections (24%)
- Neurological complications (10%)
- Cancer (8%)
- Life expectancy reduced by 10-15 years in patients progressing to ESRD
- 5-year survival on dialysis: 65-80% (better than most other causes of ESRD)
- Post-transplant outcomes generally excellent (90% 5-year graft survival)
ARPKD-Specific Complications:
- Neonatal respiratory failure due to pulmonary hypoplasia (30-40% mortality in severe cases)
- Congenital hepatic fibrosis leading to portal hypertension
- Growth retardation
- Earlier onset of hypertension
- More rapid progression to kidney failure
The multiple systemic complications of PKD necessitate a comprehensive approach to patient management, often involving multidisciplinary care teams and careful consideration of screening for extrarenal manifestations. Appropriate complication monitoring and management can significantly improve quality of life and outcomes for affected individuals.
7. Diagnosis & Testing
The diagnosis of Polycystic Kidney Disease involves a combination of imaging studies, clinical evaluation, and genetic testing. Early and accurate diagnosis is essential for appropriate management and family screening.
Clinical Evaluation:
Medical History and Physical Examination:
- Family history assessment (crucial for ADPKD)
- Evaluation of symptoms (pain, hematuria, hypertension)
- Abdominal examination for kidney or liver enlargement
- Blood pressure measurement
- Assessment for extrarenal manifestations
Laboratory Assessments:
- Kidney function tests:
- Serum creatinine
- Estimated glomerular filtration rate (eGFR)
- Blood urea nitrogen (BUN)
- Urinalysis (for hematuria, proteinuria, signs of infection)
- Complete blood count (anemia may indicate advanced disease)
- Liver function tests
- Electrolyte panel
- Urine concentration tests (early defect in PKD)
- Kidney function tests:
Imaging Techniques:
Ultrasonography:
- First-line and most widely used imaging modality
- Advantages:
- Non-invasive
- No radiation
- Relatively inexpensive
- Widely available
- Unified diagnostic criteria by age for ADPKD:
- Age 15-39: ≥3 cysts (total bilateral)
- Age 40-59: ≥2 cysts in each kidney
- Age ≥60: ≥4 cysts in each kidney
- Limitations:
- Operator-dependent
- Less sensitive for small cysts (<1cm)
- Limited utility in early disease
Computed Tomography (CT):
- Higher sensitivity than ultrasound
- Provides detailed anatomical information
- Useful for:
- Complications (bleeding, infection)
- Surgical planning
- Evaluation of kidney stones
- Limitations:
- Radiation exposure
- Contrast risks in impaired kidney function
- Higher cost
Magnetic Resonance Imaging (MRI):
- Gold standard for kidney volume assessment
- Highest sensitivity for detecting small cysts
- Enables accurate measurement of total kidney volume (TKV)
- T2-weighted images excellent for cyst visualization
- Used to monitor disease progression in clinical trials
- Limitations:
- Higher cost
- Limited availability
- Time-consuming
- Contraindicated with certain implants
MR Angiography:
- For screening of intracranial aneurysms in high-risk patients
- Non-invasive alternative to conventional angiography
- Recommended for:
- Patients with family history of aneurysm/hemorrhage
- High-risk occupations
- Prior to major surgeries
- Symptoms suggesting aneurysm
Genetic Testing:
Next-Generation Sequencing (NGS):
- Comprehensive analysis of PKD1, PKD2, and other related genes
- Used for:
- Confirmation of diagnosis in uncertain cases
- Presymptomatic diagnosis in at-risk individuals
- Family planning
- Potential kidney donors from affected families
- Detects approximately 90% of disease-causing mutations
- Challenges:
- PKD1 gene complexity (large gene with multiple pseudogenes)
- Variants of uncertain significance
- Cost and insurance coverage
Linkage Analysis:
- Used for large families when direct mutation testing is unavailable
- Requires multiple affected family members
- Less commonly used with advances in direct sequencing
Preimplantation Genetic Diagnosis (PGD):
- Used with in vitro fertilization
- Allows selection of embryos without PKD mutations
- Option for families wishing to prevent disease transmission
Prenatal Diagnosis:
Ultrasonography:
- Can detect enlarged, echogenic kidneys in ARPKD and severe ADPKD
- Limited sensitivity before 24 weeks of gestation
Amniocentesis or Chorionic Villus Sampling:
- Enables genetic testing of fetal cells
- Used when family mutation is known
- Carries small procedural risk
Screening Recommendations:
For At-Risk Individuals (ADPKD):
- First-degree relatives of affected individuals
- Presymptomatic screening usually offered after age 18-21
- Earlier screening if results would alter management (e.g., sports participation)
- Ultrasound is initial screening modality
- Negative ultrasound before age 30 does not exclude the disease
For ARPKD Carriers:
- Parental carrier testing if a child is affected
- Prenatal diagnosis in subsequent pregnancies if desired
Diagnostic Challenges:
Differential Diagnosis:
- Simple kidney cysts (common with aging)
- Tuberous sclerosis complex
- Von Hippel-Lindau disease
- Other ciliopathies
- Medullary sponge kidney
- Acquired cystic kidney disease (in patients with existing kidney disease)
Atypical Presentations:
- Very mild PKD (especially PKD2 mutations)
- De novo mutations without family history
- Mosaicism causing variable expressivity
- Contiguous gene syndromes
Emerging Biomarkers:
- Urinary exosomes containing polycystins
- Proteomics and metabolomics profiles
- MicroRNAs
- Still investigational, not in routine clinical use
Early diagnosis of PKD allows for:
- Appropriate monitoring and intervention
- Family planning
- Lifestyle modifications to slow progression
- Treatment of hypertension and other complications
- Preparation for eventual kidney replacement therapy if needed
The diagnostic approach should be tailored to the individual situation, considering family history, age, and the purpose of testing (confirmation of diagnosis, family screening, or prenatal diagnosis).
8. Treatment Options
Management of Polycystic Kidney Disease has evolved significantly, with approaches ranging from supportive care to disease-modifying therapies and renal replacement options. Treatment strategies must address both the primary disease process and its various complications.
Disease-Modifying Treatments:
Vasopressin V2 Receptor Antagonists:
Tolvaptan (Jynarque/Samsca):
- First FDA-approved treatment for ADPKD (2018)
- Slows kidney growth and GFR decline by approximately 30%
- Mechanism: Blocks vasopressin’s effect on cAMP production
- Target population: Adults with rapidly progressing disease
- Side effects: Polyuria, thirst, liver enzyme abnormalities
- Requires liver function monitoring
- Significant cost (>$10,000/month)
Practical considerations:
- Patient selection based on age, kidney function, and progression risk
- Contraindicated in advanced CKD (eGFR <25 ml/min)
- Requires significant water intake (2-3 liters daily minimum)
Somatostatin Analogues:
- Octreotide, Lanreotide:
- Reduce cAMP production in liver and kidney
- Evidence for slowing both kidney and liver cystic disease
- More studied for liver cysts than kidney cysts
- Not FDA-approved for PKD, but used off-label
- Side effects: Gallstones, GI disturbances, glucose abnormalities
- Monthly injections
- Octreotide, Lanreotide:
Management of Hypertension:
Angiotensin-Converting Enzyme Inhibitors (ACEIs) and Angiotensin Receptor Blockers (ARBs):
- First-line agents for PKD-related hypertension
- Target blood pressure: <130/80 mmHg in most patients
- May slow progression beyond blood pressure effects
- Side effects: Cough (ACEIs), angioedema, hyperkalemia
Other Antihypertensive Agents:
- Calcium channel blockers
- Diuretics (used cautiously)
- Beta-blockers
- Often require multiple agents for control
Pain Management:
Acute Pain Episodes:
- NSAIDs (short-term use with caution)
- Opioid analgesics for severe episodes
- Heat therapy
Chronic Pain:
- Acetaminophen
- Tricyclic antidepressants
- Gabapentin/pregabalin for neuropathic components
- Pain psychology approaches
- Avoidance of long-term opioids when possible
Interventional Procedures:
- Cyst aspiration and sclerotherapy for dominant symptomatic cysts
- Cyst decortication (laparoscopic or open)
- Nerve blocks
- Splanchnic nerve ablation
- Rarely, nephrectomy for intractable pain
Management of Liver Cysts:
Conservative Management:
- Avoidance of estrogens (can stimulate growth)
- Symptomatic treatment
Interventional Procedures:
- Aspiration and sclerotherapy for dominant cysts
- Fenestration procedures (laparoscopic or open)
- Segmental hepatic resection
- Rarely, liver transplantation for massive hepatomegaly
Renal Replacement Therapy:
Dialysis:
- Hemodialysis
- Peritoneal dialysis (may be challenging with very large kidneys)
- Initiated at similar GFR thresholds as other kidney diseases
- Outcomes generally better than other causes of ESRD
Kidney Transplantation:
- Treatment of choice for ESRD in PKD
- Excellent outcomes (equal or better than other indications)
- Considerations:
- Native nephrectomy sometimes needed for space
- Screening for cerebral aneurysms recommended
- Living related donors require careful evaluation to exclude PKD
- No disease recurrence in the transplanted kidney
Combined Liver-Kidney Transplantation:
- For selected patients with both severe polycystic liver and kidney disease
Surgical Interventions:
Nephrectomy:
- Indications:
- Recurrent serious infections
- Intractable pain
- Suspicion of malignancy
- Space issues before transplantation
- Recurrent significant bleeding
- Approaches:
- Laparoscopic (preferred when feasible)
- Open surgical
- Indications:
Cyst Decompression Procedures:
- Used for symptom control
- Not proven to preserve kidney function
- Higher recurrence rates than nephrectomy
Management of Complications:
Urinary Tract Infections:
- Prolonged antibiotic courses often needed
- Cyst infections may require lipophilic antibiotics
Kidney Stones:
- Medical management based on stone composition
- Extracorporeal shock wave lithotripsy
- Ureteroscopy
- Percutaneous approaches
Intracranial Aneurysms:
- Screening with MR angiography for high-risk patients
- Management options:
- Observation for small (<7mm), asymptomatic aneurysms
- Neurosurgical clipping
- Endovascular coiling
Lifestyle and Supportive Measures:
Dietary Recommendations:
- Adequate hydration (2.5-3L daily)
- Sodium restriction (<2.3g daily)
- Moderate protein intake
- Avoid grapefruit juice (interferes with tolvaptan)
- Limited caffeine
Physical Activity:
- Regular exercise encouraged
- Avoidance of high-impact or contact sports with large kidneys
- Adaptation based on symptoms and kidney size
Psychosocial Support:
- Genetic counseling
- Support groups
- Mental health services
Emerging and Investigational Therapies:
mTOR Inhibitors:
- Sirolimus, everolimus
- Initial promising results not confirmed in larger trials
- May still have role in selected patients
Tyrosine Kinase Inhibitors:
- Tesevatinib and other multi-kinase inhibitors
- Target epidermal growth factor receptor (EGFR) signaling
- In clinical trials
HDAC Inhibitors:
- Valproic acid, trichostatin A
- Alter gene expression patterns
- Preclinical and early clinical investigation
Metformin:
- AMPK activation
- Potential anti-cystic effects
- Clinical trials ongoing
Bardoxolone Methyl:
- Anti-inflammatory and antioxidant effects
- Phase 2 trials underway
Gene Therapy Approaches:
- Still in preclinical development
- Challenges include delivery to kidney cells and gene size
The management of PKD requires a comprehensive, multidisciplinary approach tailored to the individual patient’s disease manifestations, progression rate, complications, and preferences. Early intervention, particularly blood pressure control and potentially disease-modifying therapy, offers the best opportunity to preserve kidney function and quality of life.
9. Prevention & Precautionary Measures
As a genetic disorder, Polycystic Kidney Disease cannot be completely prevented in those who have inherited the causative mutations. However, several approaches can prevent disease transmission, slow progression, and minimize complications.
Genetic Counseling and Family Planning:
Genetic Counseling:
- Essential for affected individuals and families
- Explains inheritance patterns and recurrence risks
- Discusses testing options for family members
- Provides psychological support for decision-making
Preimplantation Genetic Diagnosis (PGD):
- Used with in vitro fertilization (IVF)
- Embryos are tested for PKD mutations before implantation
- Only unaffected embryos are transferred
- Prevents disease transmission to offspring
- Considerations:
- High cost
- Limited insurance coverage
- Ethical dimensions
- Success rates similar to standard IVF
Prenatal Diagnosis:
- Options include:
- Chorionic villus sampling (10-13 weeks)
- Amniocentesis (15-20 weeks)
- Allows for informed reproductive decisions
- Less commonly utilized than in the past due to PGD availability
- Options include:
Carrier Testing:
- For ARPKD, identifies parents carrying single mutations
- Essential for recurrence risk assessment
- Particularly important in consanguineous families
Measures to Slow Disease Progression:
Blood Pressure Management:
- Rigorous control to target <130/80 mmHg
- Regular home monitoring
- Lifestyle measures plus pharmacotherapy
- Early initiation of ACEIs or ARBs
- May slow cyst growth independent of blood pressure effects
Hydration and Vasopressin Suppression:
- High water intake (2.5-3L daily) to suppress vasopressin
- Evenly distributed water consumption throughout day and evening
- Avoiding concentrated urine (target urine osmolality <280 mOsm/kg)
- Tolvaptan for eligible patients with rapidly progressive disease
Dietary Modifications:
- Sodium restriction (<2.3g daily)
- Reduces blood pressure
- May directly slow cyst growth
- Enhances effectiveness of RAAS blockade
- Moderate protein intake (0.8-1.0 g/kg/day)
- Reduces hyperfiltration
- Minimizes uremic toxin load
- Limited caffeine intake
- Reduces cAMP stimulation
- Adequate potassium intake
- Sodium restriction (<2.3g daily)
Lifestyle Factors:
- Maintenance of healthy body weight
- Regular physical activity adapted to kidney size
- Smoking cessation
- Accelerates kidney function decline
- Increases cardiovascular risk
- Alcohol in moderation
Metabolic Control:
- Management of comorbid diabetes
- Treatment of dyslipidemia
- Avoidance of hyperuricemia and gout
Prevention and Management of Complications:
Urinary Tract Infection Prevention:
- Adequate hydration
- Prompt treatment of symptomatic episodes
- Consideration of prophylaxis in recurrent cases
- Perineal hygiene measures
Kidney Stone Prevention:
- High fluid intake
- Dietary modifications based on stone type
- Medical therapy when indicated
- Regular monitoring in those with history of stones
Intracranial Aneurysm Screening:
- Recommended for:
- Patients with family history of aneurysm/subarachnoid hemorrhage
- Those with high-risk occupations
- Patients with previous aneurysms
- Before major elective surgery
- When symptoms suggest possible aneurysm
- Typically performed with MR angiography
- Interval depends on initial findings (3-5 years if negative)
- Recommended for:
Cardiac Surveillance:
- Echocardiography to assess for valve abnormalities
- Standard cardiovascular risk factor management
- Enhanced attention to lipid management
Liver Cyst Monitoring:
- Periodic liver imaging
- Avoidance of estrogen therapy in women with extensive hepatic cysts
- Prompt evaluation of liver-related symptoms
Education and Supportive Measures:
Patient Education:
- Disease nature and progression
- Importance of blood pressure control
- Recognition of complications requiring attention
- Available treatment options
- Genetic implications
Regular Monitoring:
- Kidney function (eGFR, creatinine)
- Blood pressure
- Urinalysis
- Imaging to track kidney size/volume
- Screening for extrarenal manifestations
Psychosocial Support:
- Addressing anxiety and depression
- Coping strategies for chronic illness
- Support groups (in-person and online)
- Family system approaches
Vocational Considerations:
- Adaptation of work environment as needed
- Disability management when appropriate
- Career planning considering disease trajectory
Advance Care Planning:
- Discussion of renal replacement options
- Transplant evaluation timing
- Documentation of care preferences
Special Populations:
Children with ARPKD:
- Aggressive management of hypertension
- Growth monitoring and nutritional support
- Developmental assessment
- Management of congenital hepatic fibrosis
Women of Childbearing Age:
- Preconception counseling
- Medication adjustments before pregnancy (discontinuation of certain agents)
- Close monitoring during pregnancy
- Management of pregnancy-related hypertension
Elderly Patients:
- Balanced approach to blood pressure management
- Consideration of comorbidities in treatment decisions
- Conservative management options when appropriate
While PKD cannot be prevented entirely, the comprehensive approach outlined above can significantly alter the disease course, delay progression to kidney failure, reduce complications, and improve quality of life. The growing understanding of pathophysiology and risk factors has led to more targeted preventive strategies, with promising results from early intervention studies.
10. Global & Regional Statistics
Polycystic Kidney Disease represents a significant global health burden, with notable variations in prevalence, management, and outcomes across different regions and healthcare systems.
Global Prevalence and Epidemiology:
ADPKD Prevalence:
- Global prevalence: 1:400 to 1:1,000 individuals
- Estimated 12.5 million people affected worldwide
- No significant ethnic or racial predilection
- Gender distribution: Equal in males and females
- Accounts for 5-10% of all cases requiring renal replacement therapy
ARPKD Prevalence:
- Much rarer: 1:20,000 to 1:40,000 live births
- Higher in populations with increased consanguinity
- Carrier frequency: Approximately 1:70
- Responsible for 5-8% of pediatric end-stage renal disease cases
Genetic Distribution:
- PKD1 mutations: ~78% of ADPKD cases
- PKD2 mutations: ~15% of ADPKD cases
- Other genes (GANAB, DNAJB11, etc.): ~7% of cases
- Mutation rates relatively consistent across populations
Regional Variations and Statistics:
North America:
- United States:
- Approximately 500,000-600,000 affected individuals
- ADPKD accounts for 5% of ESRD cases
- Mean age at ESRD: 53 years
- Transplantation rate: 75-80% of eligible patients
- Annual healthcare costs: $50,000-$100,000 per patient with advanced disease
- Canada:
- Similar prevalence to US
- Higher transplantation rates (>80% of eligible patients)
- Universal healthcare facilitates earlier intervention
- United States:
Europe:
- Western Europe:
- Similar prevalence to North America
- Higher utilization of genetic testing
- Earlier adoption of tolvaptan therapy
- Regional registries providing valuable epidemiological data
- Eastern Europe:
- More variable access to specialized care
- Lower transplantation rates
- Later diagnosis on average
- Western Europe:
Asia:
- Japan:
- Prevalence approximately 1:4,000
- Pioneer in tolvaptan development and use
- Earlier age at diagnosis due to systematic screening
- High imaging availability
- China:
- Estimated 3-4 million affected individuals
- Growing recognition and diagnosis
- Variable access to specialized care between urban and rural areas
- Lower transplantation rates
- India:
- Limited epidemiological data
- Later diagnosis and treatment
- Cost barriers to specialized care
- Lower rates of renal replacement therapy
- Japan:
Middle East:
- Higher ARPKD rates in some countries due to consanguinity
- ADPKD prevalence similar to global averages
- Emerging specialized centers in wealthy Gulf states
- Regional variations in access to genetic testing
Africa:
- Limited epidemiological data
- Significant underdiagnosis likely
- Restricted access to renal replacement therapy
- Very low transplantation rates in most countries
Latin America and Caribbean:
- Similar genetic distribution to North America
- Variable healthcare access
- Dialysis more common than transplantation
- Growth in specialized PKD clinics in major urban centers
Mortality and Survival Rates:
Overall Mortality:
- Standardized mortality ratio: 1.6-3.2 times general population
- Higher in younger patients and those reaching ESRD
- Cardiovascular disease: leading cause of death (35-40%)
ESRD and Renal Replacement Therapy:
- Proportion reaching ESRD by age 80: 50-75%
- Mean age at ESRD: ~58 years (varies by genotype)
- Regional variations in ESRD onset:
- Europe/North America: 55-60 years
- Japan: 52-54 years
- Developing regions: Limited data, likely earlier
Dialysis Outcomes:
- 5-year survival on dialysis: 65-80%
- Better than other causes of ESRD (diabetic nephropathy: 35-40%)
- Regional variations in dialysis mortality:
- North America: 15-20% annual mortality
- Europe: 10-15% annual mortality
- Developing regions: 20-30% annual mortality
Transplantation Outcomes:
- 1-year graft survival: 92-96%
- 5-year graft survival: 85-90%
- Better outcomes than most other causes of ESRD
- Patient survival better than on dialysis (50-70% reduction in mortality)
Healthcare Utilization and Economic Impact:
Healthcare Resource Utilization:
- Hospitalization rates: 0.2-0.5 admissions per patient per year
- Emergency department visits: 0.5-1.0 per patient per year
- Imaging studies: 1-3 per patient per year
- Specialist visits: 2-6 per patient per year
Economic Burden:
- Global economic burden: $7.5-10 billion annually
- Direct medical costs per patient:
- Early disease: $4,000-$10,000 annually
- Advanced disease: $30,000-$50,000 annually
- ESRD care: $70,000-$100,000 annually
- Indirect costs (lost productivity): $3-5 billion annually
- Regional variations in cost:
- Highest in US
- Intermediate in Europe and Canada
- Lower in developing regions (but higher as percentage of GDP/capita)
Treatment Availability:
- Tolvaptan approval and access:
- Approved in 30+ countries
- Coverage/reimbursement varies significantly
- Cost barrier in many regions
- Transplantation availability:
- Highest in Northern Europe, Canada, and US
- Intermediate in most developed countries
- Limited in developing regions
- Living donor programs variable by region
- Tolvaptan approval and access:
Current Trends:
Earlier Diagnosis:
- Increasing through family screening
- Growing use of genetic testing
- Incidental findings on imaging for other conditions
Treatment Patterns:
- Expanding use of disease-modifying therapies
- More aggressive blood pressure management
- Earlier transplantation evaluation
- Growth in specialized PKD clinics
Research Focus:
- Greater participation in international registries
- Expansion of clinical trials to diverse populations
- Biomarker development for personalized treatment
- Increased attention to health-related quality of life
The global landscape of PKD continues to evolve, with improvements in diagnosis, management, and outcomes in many regions but persistent disparities in access to specialized care and advanced therapies. International collaboration through registries and research networks is helping to address knowledge gaps and develop more effective management strategies.
11. Recent Research & Future Prospects
The field of Polycystic Kidney Disease research has experienced remarkable progress in recent years, with advances in understanding disease mechanisms leading to novel therapeutic approaches and promising future directions.
Recent Major Research Advances:
Pathophysiological Insights:
- Ciliary Biology: Enhanced understanding of primary cilia as cellular antennae and their role in PKD
- Cellular Crosstalk: Identification of complex interactions between cystic epithelial cells and surrounding matrix/interstitium
- Metabolic Reprogramming: Discovery of Warburg-like metabolic alterations in cyst-lining cells
- Epigenetic Modifications: Recognition of their role in cyst formation and progression
- Inflammasome Activation: Emerging evidence for inflammatory pathways in cystogenesis
Biomarker Development:
- Imaging Biomarkers: Validation of total kidney volume (TKV) as a prognostic marker
- Urinary Biomarkers: Identification of exosomal markers (polycystins, microRNAs)
- Plasma Proteomics: Discovery of circulating protein signatures of rapid progression
- Genetic Modifiers: Identification of variants affecting disease severity
Clinical Trial Advances:
- TEMPO 3:4 and REPRISE Trials: Demonstrated tolvaptan’s efficacy in slowing progression
- DIPAK-1 Trial: Evaluated lanreotide for combined kidney/liver cystic disease
- HALT-PKD: Defined optimal blood pressure targets and RAAS blockade approaches
- MDT-PWV Trial: Explored metformin’s potential in ADPKD
Current Treatment Development:
Novel Pharmacological Approaches:
Glucosylceramide Synthase Inhibitors:
- Venglustat (Sanofi) in Phase 3 trials
- Targets glycosphingolipid pathway
- Potential benefits for both kidney and liver cysts
AMPK Activators:
- Metformin in Phase 2/3 trials
- Modulates cellular energy metabolism
- May reprogram cystic epithelial metabolism
Second-Generation V2 Receptor Antagonists:
- Lixivaptan in Phase 3 trials
- Potentially improved hepatic safety profile
- Similar mechanism to tolvaptan
HDAC Inhibitors:
- Valproic acid repurposing
- Modulates epigenetic regulation
- Preclinical evidence for cyst reduction
Anti-inflammatory Approaches:
- NF-κB inhibitors
- IL-1β antagonists
- Targeting inflammation-cyst growth cycle
Combination Therapies:
- V2 antagonist + mTOR inhibitor combinations
- Somatostatin analogs + metformin
- Tyrosine kinase inhibitors + HDAC inhibitors
- Multi-target approaches to address complex pathophysiology
Novel Delivery Systems:
- Kidney-targeted drug delivery platforms
- Nanoparticle formulations for PKD drugs
- Sustained-release implants
- Enhanced cyst penetration technologies
Genetic and Regenerative Approaches:
Gene Therapy:
CRISPR/Cas9 Approaches:
- In vivo gene editing to correct PKD mutations
- Ex vivo correction of patient-derived cells
- Technical challenges include delivery to kidney cells
RNA Therapeutics:
- Antisense oligonucleotides for specific mutations
- RNA interference targeting disease pathways
- microRNA modulation
Stem Cell Strategies:
- Patient-derived induced pluripotent stem cells (iPSCs)
- Gene-corrected autologous cell therapy
- Bioengineered kidney tissues
- Stem cell-derived extracellular vesicles as therapy
Organ Regeneration:
- Bioartificial kidney devices
- 3D-printed kidney structures
- Organoid models for personalized drug testing
- Xenotransplantation approaches
Emerging Clinical Strategies:
Precision Medicine:
- Genotype-phenotype correlation for personalized prognosis
- Biomarker-guided treatment selection
- Pharmacogenomic approaches to predict drug response
- Risk stratification tools for clinical decision-making
Innovative Clinical Trial Designs:
- Adaptive platform trials testing multiple agents
- Basket trials grouping mechanistically similar diseases
- Enrichment strategies targeting high-risk patients
- Novel endpoints beyond traditional kidney function measures
Digital Health Integration:
- Remote monitoring of disease parameters
- Smartphone-based symptom tracking
- Patient-reported outcome collection
- Machine learning for progression prediction
Major Research Initiatives and Collaborations:
HALT Progression of Polycystic Kidney Disease (HALT PKD) Network:
- Multicenter collaborative research program
- Focus on hypertension management and disease progression
- Landmark studies defining blood pressure management
Polycystic Kidney Disease Outcomes Consortium (PKDOC):
- FDA/EMA-qualified total kidney volume as prognostic biomarker
- Standardized imaging and biomarker protocols
- Accelerated clinical trial development
ADPKD Registry Studies:
- ADPKD Registry (US-based)
- ADPedKD (pediatric ADPKD registry)
- EuroCYST Initiative (European ADPKD Consortium)
- Global harmonization of data collection
PKD Foundation Research Network:
- Connecting researchers globally
- Targeted research grant programs
- Acceleration of translational research
Future Research Horizons:
Systems Biology Approaches:
- Multi-omics integration (genomics, proteomics, metabolomics)
- Network analysis of disease pathways
- Computational modeling of cyst growth dynamics
- Virtual patient simulations for treatment optimization
Artificial Intelligence Applications:
- Automated TKV measurement from imaging
- Machine learning prediction of rapid progressors
- Deep learning analysis of kidney biopsy images
- Natural language processing of medical records
Novel Therapeutic Targets:
- Extracellular vesicle signaling
- Mitochondrial dysfunction
- Autophagy modulation
- Fluid shear stress sensing
- Circadian rhythm regulation
Preventive Strategies:
- Early intervention before cyst formation
- Pre-symptomatic treatment of at-risk individuals
- Kidney-protective protocols from childhood
- Novel genetic approaches to prevent disease transmission
The future of PKD research appears promising, with multiple complementary approaches advancing simultaneously. The convergence of improved understanding of disease mechanisms, novel therapeutic targets, precision medicine approaches, and innovative clinical trial designs offers hope for transformative improvements in patient outcomes. While challenges remain, particularly in translating genetic approaches to clinical application, the momentum of research suggests that significantly improved management options may become available in the next decade.
12. Interesting Facts & Lesser-Known Insights
The world of Polycystic Kidney Disease contains many fascinating aspects that are not widely recognized, along with several misconceptions that deserve clarification. These insights provide a deeper understanding of this complex condition.
Historical and Scientific Curiosities:
Evolution of PKD Genes:
- PKD genes are highly conserved across species, from fish to humans
- Similar genes exist in simple organisms like fruit flies, suggesting fundamental biological importance
- Polycystins likely evolved over 600 million years ago
- May have originally served as mechanosensors in primitive organisms
PKD in Famous Historical Figures:
- Several historical figures are speculated to have had PKD, including King Ferdinand of Romania
- The composer Franz Schubert may have suffered from complications of undiagnosed PKD
- The painter Amedeo Modigliani’s kidney disease has been retrospectively suggested as possible PKD
Scientific Milestones:
- PKD1 was one of the first genes identified through positional cloning, a groundbreaking technique
- The polycystin proteins were among the first recognized components of primary cilia
- PKD research helped establish the entire field of ciliopathies (diseases of primary cilia)
- ADPKD was one of the first adult diseases recognized to have embryonic developmental pathways reactivated
Unusual Clinical Features:
Extrarenal Manifestations Beyond the Commonly Known:
- Arachnoid membrane cysts: 8-12% of ADPKD patients have these brain cysts
- Pericardial cysts: Rare but documented in PKD patients
- Spinal meningeal cysts: Can cause radicular symptoms
- Seminal vesicle cysts: Present in 40% of men with ADPKD but rarely diagnosed
- Retinal abnormalities: Present in up to 20% of patients but usually asymptomatic
Cognitive and Neuropsychological Aspects:
- Subtle cognitive differences observed in some studies (processing speed, executive function)
- Higher prevalence of anxiety disorders (30-40%) compared to general population
- Unique psychological challenges of living with a predictable but variable genetic disease
- “Genetic guilt” phenomenon in parents who have transmitted the disease
Reproductive and Hormonal Connections:
- Polycystic ovary syndrome (PCOS) more common in women with ADPKD
- Increased risk of ectopic pregnancy due to ciliary dysfunction in fallopian tubes
- Sperm motility issues in some men with ADPKD
- Estrogen receptors present on cyst-lining cells, explaining female predominance of liver cysts
Surprising Scientific Insights:
Ciliary Connection:
- Primary cilia on kidney cells function as “cellular antennae”
- These tiny hair-like projections (3-5 μm) sense fluid flow
- PKD was critical to discovering the importance of primary cilia, once thought to be vestigial
- The connection links PKD to other ciliopathies (Bardet-Biedl syndrome, nephronophthisis)
Two-Hit Hypothesis:
- Each kidney cyst likely originates from a single cell
- Requires germline mutation plus “second hit” (somatic mutation)
- Explains why only 1% of nephrons develop cysts despite all cells carrying the mutation
- Similar mechanism to some cancers (though PKD cysts are benign)
Metabolic Peculiarities:
- Cystic cells exhibit the “Warburg effect” (aerobic glycolysis) like cancer cells
- Kidney cysts consume glucose differently than normal kidney tissue
- Cyst fluid contains unique metabolites and signaling molecules
- Cystic cells show alterations in fatty acid metabolism
Somatic Mosaicism:
- Some patients have mutations in only a portion of their cells
- Can lead to asymmetric disease (one kidney much more affected)
- Explains some cases of sporadic PKD without family history
- Challenging to detect with standard genetic testing
Debunking Myths and Misconceptions:
Myth: All PKD patients develop kidney failure
- Reality: Up to 30% of patients with PKD2 mutations never reach kidney failure
- Many patients die of unrelated causes before developing ESRD
- Significant variability exists even with identical mutations
Myth: PKD only affects the kidneys
- Reality: It’s a systemic disorder affecting multiple organ systems
- Some patients have predominant liver rather than kidney disease
- Vascular manifestations can be life-threatening independent of kidney function
Myth: Nothing can be done to slow PKD progression
- Reality: Multiple evidence-based interventions now exist
- Blood pressure control, lifestyle modifications, and targeted therapies can significantly alter the disease course
- Early intervention appears particularly beneficial
Myth: PKD patients should avoid physical activity
- Reality: Regular exercise is beneficial for most patients
- Only contact sports pose significant risk with very enlarged kidneys
- Physical activity helps control blood pressure and maintain cardiovascular health
Myth: Kidney cysts can become cancerous
- Reality: Despite similarities to cancer cells, PKD cysts remain benign
- No increased risk of renal cell carcinoma in PKD patients
- Different molecular mechanisms from cancerous growths
Impact on Specific Populations and Professions:
Occupational Considerations:
- Military Service: Most branches exclude those with diagnosed PKD
- Commercial Pilots: Restrictions based on kidney function and aneurysm risk
- Professional Athletes: Potential limitations for contact sports
- Commercial Diving: Often restricted due to potential kidney injury risks
Pregnancy and Family Planning Impacts:
- 80% of pregnancies in women with PKD are successful
- Higher rates of preeclampsia (25-30%)
- Psychological complexity of family planning decisions
- Gender differences in how disease transmission risk affects family planning choices
Cultural and Ethnic Aspects:
- Cultural variations in acceptance of genetic testing
- Different approaches to family screening across cultures
- Variable attitudes toward preimplantation genetic diagnosis
- Consanguinity increases ARPKD risk in some communities
Practical Insights for Daily Living:
Subtle Early Signs Often Missed:
- Mild hypertension often the earliest detectable sign
- Nocturia (nighttime urination) frequently attributed to other causes
- Subtle lower back discomfort often dismissed
- Fatigue and reduced stamina may precede laboratory abnormalities
Unexpected Symptom Triggers:
- Air travel can exacerbate kidney pain due to gas expansion in cysts
- Roller coasters and high-impact activities may trigger cyst bleeding
- Caffeine and high-sodium meals can acutely worsen symptoms
- Hormonal fluctuations may affect cyst-related pain in women
Insurance and Financial Planning:
- Life insurance challenges for diagnosed patients
- Importance of securing coverage before genetic testing
- Long-term financial planning for potential disability
- Navigating health insurance with a pre-existing condition
Psychological Adaptation:
- “Genetic anticipation” phenomenon (earlier diagnosis in each generation due to awareness)
- “Survivor guilt” in unaffected family members
- Unique challenges of “presymptomatic” diagnosis
- Value of peer support from others with PKD
The rich tapestry of lesser-known aspects of PKD reflects its complexity as both a medical condition and a lived experience. These insights not only enhance understanding but can also inform more holistic approaches to patient care and support, recognizing the multifaceted nature of living with a progressive genetic condition.