⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
Comprehensive Report on Spinal Muscular Atrophy
1. Overview
What is Spinal Muscular Atrophy?
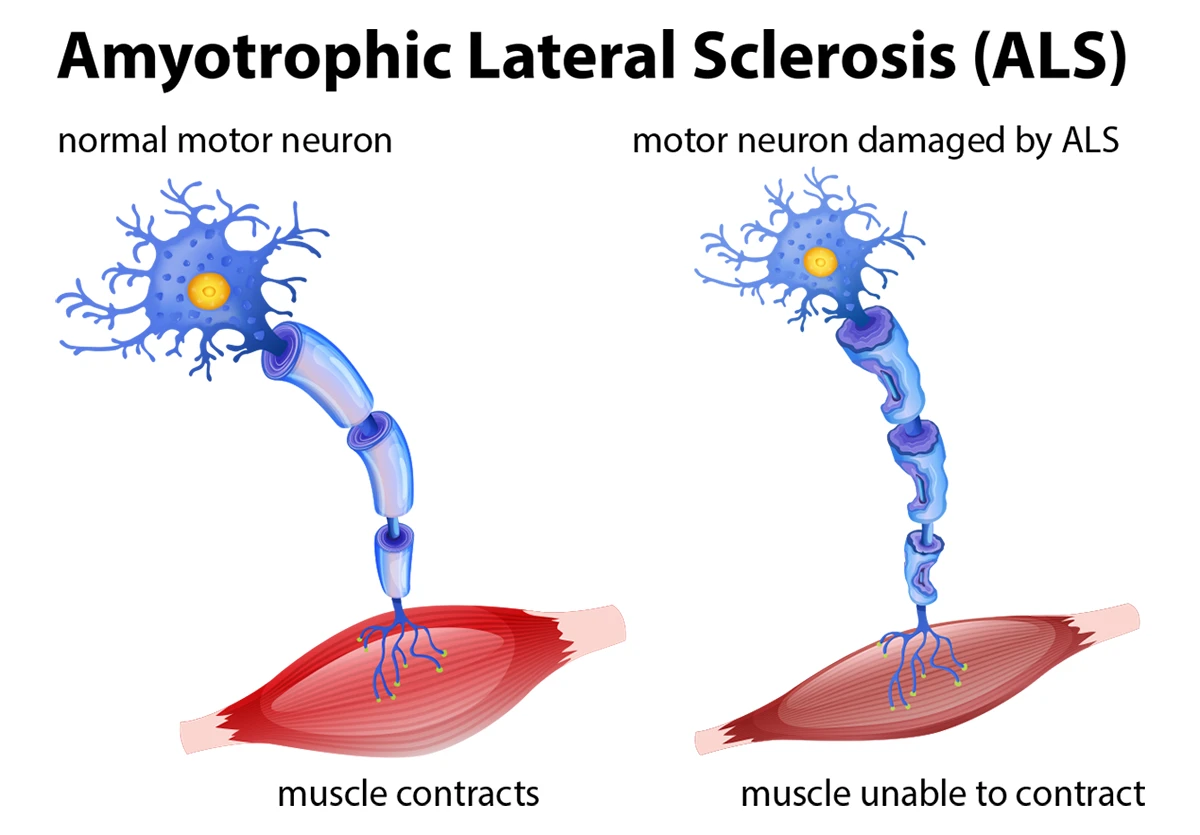
Spinal Muscular Atrophy (SMA) is a rare, genetic neuromuscular disorder characterized by the degeneration of motor neurons in the anterior horn of the spinal cord and the brain stem nuclei. This degeneration leads to progressive muscle weakness and atrophy, predominantly affecting the proximal muscles.
SMA is caused by a deficiency of the survival motor neuron (SMN) protein, which is critical for the functioning and survival of motor neurons. Without sufficient SMN protein, motor neurons gradually die, resulting in the inability to transmit signals between the brain and the muscles, ultimately leading to muscle weakness and atrophy.
Detailed Classification
SMA is classified into several types based on the age of onset and severity of symptoms:
Type 0 (Prenatal SMA): The most severe form, present before birth with decreased fetal movement. Infants are born with severe weakness, contractures, and respiratory failure. Survival beyond a few weeks is rare.
Type I (Werdnig-Hoffmann Disease): Onset within the first 6 months of life. Infants never achieve the ability to sit independently. Without intervention, life expectancy is typically less than 2 years due to respiratory failure.
Type II (Dubowitz Disease): Onset between 6-18 months. Children can sit independently but never stand or walk without assistance. Life expectancy varies but can extend into adulthood with proper care.
Type III (Kugelberg-Welander Disease): Onset after 18 months. Children can stand and walk independently, though they may lose this ability over time. Normal lifespan with variable degrees of disability.
Type IV (Adult-Onset SMA): Rare form with onset in adulthood (typically after age 30). Presents with mild to moderate muscle weakness, primarily affecting the proximal muscles. Normal lifespan with slowly progressive disability.
Affected Body Parts/Organs
SMA primarily affects the voluntary muscles used for activities like crawling, walking, head and neck control, and swallowing. The specific systems affected include:
Musculoskeletal System:
- Proximal muscles (shoulders, hips, and trunk) more severely affected than distal muscles
- Symmetrical muscle weakness and atrophy
- Sparing of facial muscles in most cases
- Progressive scoliosis and joint contractures
Respiratory System:
- Intercostal muscle weakness
- Diaphragmatic breathing pattern
- Impaired cough reflex
- Increased susceptibility to respiratory infections
Gastrointestinal System:
- Bulbar muscle involvement leading to difficulties in chewing and swallowing
- Increased risk of aspiration
- Gastroesophageal reflux
- Nutritional challenges
Nervous System:
- Degeneration of alpha motor neurons in the anterior horn of the spinal cord
- Preservation of cognitive and sensory functions
- Variable involvement of brainstem motor nuclei
- Occasional autonomic nervous system involvement in severe forms
Prevalence and Significance
SMA is one of the leading genetic causes of infant mortality worldwide. Key epidemiological aspects include:
- Incidence: Approximately 1 in 6,000 to 1 in 10,000 live births are affected by SMA
- Carrier Frequency: About 1 in 40-50 individuals carry a single copy of the mutated gene
- Distribution: SMA affects individuals across all ethnic groups, though carrier frequencies vary by population
- Mortality: Without treatment, SMA Type I is the most common genetic cause of death in infants
- Economic Impact: The financial burden of SMA care can exceed $1 million in the first year of life for severe cases
- Healthcare Utilization: Patients with SMA have significantly higher healthcare utilization rates compared to age-matched controls
The significance of SMA extends beyond its prevalence due to:
- The devastating impact on affected individuals and their families
- The high cost of care and economic burden
- Its status as a model disease for developing genetic therapies
- Recent breakthroughs in treatment that have transformed the natural history of the disease
- The ethical and economic challenges surrounding access to these high-cost treatments
2. History & Discoveries
First Identification
The first documented descriptions of SMA emerged in the late 19th century:
1891: Austrian neurologist Guido Werdnig published the first clear clinical description of infantile SMA, documenting two brothers with progressive muscle weakness beginning in infancy.
1893: German neurologist Johann Hoffmann independently reported similar cases and provided further detailed pathological findings, noting the degeneration of anterior horn cells in the spinal cord.
1900: Werdnig and Hoffmann’s work was collectively recognized, leading to the eponym “Werdnig-Hoffmann disease” for what is now classified as SMA Type I.
1956: Erik Kugelberg and Lisa Welander described a milder, juvenile form of the disease, now known as SMA Type III.
Key Historical Figures
Several pioneers contributed significantly to our understanding of SMA:
Guido Werdnig (1844-1919): Austrian neurologist who provided the first clinical description of infantile SMA.
Johann Hoffmann (1857-1919): German neurologist who expanded on Werdnig’s observations and contributed essential pathological insights.
Eric Kugelberg (1913-1983) and Lisa Welander (1909-2001): Swedish neurologists who characterized the juvenile form of SMA.
Victor Dubowitz (1931-): British pediatric neurologist who described the intermediate form (Type II) and developed comprehensive classification systems.
Judith Melki and Team (1990s): Identified the genetic cause of SMA, mapping it to chromosome 5q and identifying the SMN1 gene.
Major Discoveries and Breakthroughs
Genetic Understanding:
- 1990: The SMA disease locus was mapped to chromosome 5q13.
- 1995: Judith Melki and colleagues identified the SMN1 gene as the SMA-determining gene, discovering that approximately 95% of SMA cases result from homozygous deletion of SMN1.
- 1997: Researchers discovered the role of SMN2, a nearly identical paralog of SMN1, as a disease modifier that influences SMA severity.
- 1999: The molecular function of the SMN protein in snRNP assembly was elucidated, explaining its critical role in RNA processing.
Clinical Classification and Management:
- 1950s-1960s: Development of initial clinical classification systems
- 1992: International SMA Consortium established standardized diagnostic criteria and classification
- 2007: Consensus statement on standards of care for SMA patients was published
- 2018: Updated care guidelines reflecting the emergence of disease-modifying therapies
Treatment Breakthroughs:
- 2000s: Preclinical development of SMN-enhancing therapeutic strategies
- December 2016: FDA approval of nusinersen (Spinraza), the first disease-modifying therapy for SMA
- May 2019: FDA approval of onasemnogene abeparvovec-xioi (Zolgensma), the first gene therapy for SMA and the most expensive drug ever approved at the time ($2.1 million per treatment)
- August 2020: FDA approval of risdiplam (Evrysdi), the first oral medication for SMA
- 2021-2024: Global expansion of treatment availability and growing experience with combination therapies
Evolution of Medical Understanding
The conceptualization and understanding of SMA have evolved significantly over time:
1890s-1950s: Descriptive Era
- Focus on clinical descriptions and pathological findings
- Classification based primarily on clinical features
- Limited understanding of pathophysiology
- Almost entirely palliative management
1950s-1980s: Clinical Refinement
- Development of more detailed classification systems
- Recognition of the spectrum of disease severity
- Improved understanding of natural history
- Development of supportive care approaches
- Emergence of multidisciplinary management
1990s: Genetic Revolution
- Identification of the genetic cause
- Understanding of SMN1 and SMN2 relationship
- Recognition of the molecular basis for clinical heterogeneity
- Development of reliable genetic testing
- Insights into the molecular functions of SMN protein
2000s-2010s: Translational Era
- Development of animal models
- Identification of therapeutic targets
- Emergence of clinical trial readiness
- Refinement of outcome measures
- Implementation of standards of care
2016-Present: Treatment Era
- FDA approval of multiple disease-modifying therapies
- Transformation of natural history of disease
- Shifting focus to early diagnosis and treatment
- Evolving standards of care incorporating new therapies
- Growing understanding of the impact of early versus late treatment
- Emergence of combination therapy approaches
This evolution reflects a remarkable journey from purely descriptive medicine to precision genetic therapy, making SMA a paradigm for genetic disease treatment and a model for the development of therapies for other genetic disorders.
3. Symptoms
Early Symptoms vs. Advanced-Stage Symptoms
Type 0 (Prenatal SMA)
- Early Signs (Prenatal):
- Decreased fetal movements
- Polyhydramnios (excess amniotic fluid)
- Joint contractures evident on ultrasound
- Sometimes associated with heart defects
- At Birth:
- Severe generalized weakness
- Respiratory distress requiring immediate ventilation
- Facial weakness affecting sucking and swallowing
- Lack of spontaneous movements
- Arthrogryposis (multiple joint contractures)
- Advanced Stage:
- Due to the extreme severity, most infants do not survive beyond a few weeks, with death typically resulting from respiratory failure.
Type I (Infantile-Onset)
- Early Symptoms (0-6 months):
- Hypotonia (“floppy infant”)
- Weak cry
- Poor head control
- Difficulty feeding and swallowing
- Paradoxical breathing (abdomen moves outward while chest moves inward during inhalation)
- Inability to lift head when prone
- “Frog-leg” posture when supine
- Advanced Symptoms (Without Treatment):
- Progressive respiratory weakness
- Recurrent respiratory infections
- Bell-shaped chest deformity
- Failure to thrive
- Bulbar dysfunction with aspiration risk
- Tongue fasciculations
- Weak cough
- Progressive difficulty handling oral secretions
- Development of early scoliosis
Type II (Later-Infantile Onset)
- Early Symptoms (6-18 months):
- Delayed motor milestones
- Ability to sit independently but inability to stand or walk without assistance
- Proximal muscle weakness
- Fine tremor of the fingers
- Decreased or absent deep tendon reflexes
- Advanced Symptoms:
- Progressive scoliosis (often requiring surgical intervention)
- Restrictive lung disease
- Development of joint contractures
- Sleep-disordered breathing
- Recurrent respiratory infections
- Potential loss of sitting ability over time
- Kyphosis or lordosis
- Hand tremors during voluntary movements
Type III (Juvenile Onset)
- Early Symptoms (>18 months):
- Delayed walking or abnormal gait
- Difficulty climbing stairs or rising from a seated position
- Proximal muscle weakness
- Frequent falls
- Muscle fatigue with activity
- Advanced Symptoms:
- Progressive weakness leading to loss of ambulation in some cases
- Scoliosis (variable severity)
- Mild respiratory involvement
- Joint contractures
- Decreased exercise tolerance
- Back pain due to postural compensation
Type IV (Adult Onset)
- Early Symptoms (>21 years, typically 30s):
- Mild proximal muscle weakness
- Difficulty rising from chairs
- Climbing stairs
- Raising arms above head
- Advanced Symptoms:
- Slowly progressive weakness
- Potential need for mobility aids in later stages
- Mild respiratory involvement in some cases
- Generally retains ability to perform activities of daily living
Common vs. Rare Symptoms
Common Symptoms Across All Types:
- Symmetrical muscle weakness, more pronounced proximally
- Absent or diminished deep tendon reflexes
- Preservation of sensation and cognitive function
- Muscle atrophy
- Fasciculations (particularly of the tongue in more severe types)
- Progressive nature of weakness
- Normal or enhanced intellectual development
Type-Specific Common Symptoms:
- Type I: “Frog-leg” positioning, paradoxical breathing, bell-shaped chest
- Type II: Fine tremor, ability to sit but not stand, progressive scoliosis
- Type III: Waddling gait, difficulty rising from floor, steppage gait
- Type IV: Mild proximal weakness with late functional impact
Rare or Less Recognized Symptoms:
- Cardiac Involvement: Primarily in severe types (0, I), including congenital heart defects, tachycardia, or cardiomyopathy
- Autonomic Dysfunction: Excessive sweating, constipation, irregular heart rate
- Metabolic Abnormalities: Altered fatty acid metabolism, glucose intolerance
- Genitourinary Issues: Bladder dysfunction, reduced fertility in males
- Skin Changes: Thin, atrophic skin with reduced subcutaneous fat
- Vascular Abnormalities: Reportedly higher risk of digital necrosis in critically ill infants
- Cognitive Effects: While cognition is typically preserved, some research suggests subtle effects on attention and working memory in some patients
- Jitteriness or Tremor: Beyond the fine tremor in Type II, some patients exhibit jitteriness that can be mistaken for seizures
Symptom Progression Over Time
Type I Progression (Untreated):
- 0-3 months: Onset of hypotonia, weak cry, feeding difficulties
- 3-6 months: Progressive weakness, difficulties handling secretions, early respiratory compromise
- 6-12 months: Significant respiratory insufficiency, potential need for ventilatory support, severe feeding difficulties often requiring tube feeding
- 12-24 months: Without intervention, respiratory failure is the usual cause of death
Type I Progression (With Treatment):
- Emerging data shows dramatically altered natural history
- Many children achieving motor milestones previously thought impossible
- Varying degrees of improvement depending on timing of treatment initiation
- Respiratory function often stabilizes or improves
- Long-term outcomes still being studied
Type II Progression:
- Infancy: Delayed motor milestones, achievement of sitting
- Early Childhood: Development of fine tremor, plateau in motor skills
- Middle Childhood: Development of scoliosis, progressive joint contractures
- Adolescence: Often marked by significant scoliosis progression during growth spurts, potential decline in respiratory function
- Adulthood: Variable stability or slow progression of weakness, respiratory function may decline gradually
Type III Progression:
- Toddler/Preschool: Achievement of walking but with abnormal gait, frequent falls
- School-Age: Increasing difficulty with stairs, running, and getting up from the floor
- Adolescence: About 50% of patients lose independent ambulation by late teens
- Young Adulthood: Those who maintain walking ability into their 20s often retain it for many years
- Later Adulthood: Gradual progression of weakness, potential need for mobility aids
Type IV Progression:
- Initial Presentation (30s-40s): Mild proximal weakness
- Middle Age: Slow progression with functional limitations
- Later Life: Usually maintains independence with adaptive equipment
Factors Affecting Progression Rate:
- SMN2 Copy Number: Higher copy numbers generally correlate with slower progression
- Age of Symptom Onset: Earlier onset generally predicts faster progression
- Nutritional Status: Malnutrition can accelerate decline
- Respiratory Management: Proactive respiratory care slows functional decline
- Access to Multidisciplinary Care: Comprehensive care improves outcomes
- Early Treatment Initiation: Earlier treatment with disease-modifying therapies shows better outcomes
- Baseline Function: Higher baseline function predicts better long-term outcomes
Understanding the spectrum and progression of symptoms is critical for clinical care, prognostication, and measuring outcomes in clinical trials. The natural history of SMA is being dramatically rewritten with the advent of effective therapies, creating a new phenotype of treated SMA that is still being characterized.
4. Causes
Biological Causes
Genetic Basis: SMA is caused by mutations in the Survival Motor Neuron 1 (SMN1) gene located on chromosome 5q13.2. This gene encodes the Survival Motor Neuron (SMN) protein, which is essential for the survival and functioning of motor neurons.
Primary Genetic Defect: In approximately 95% of SMA cases, the disease results from homozygous deletion or mutation of the SMN1 gene. Most commonly, this involves deletion of exon 7 in both copies of the gene.
SMN Protein Function: The SMN protein plays critical roles in:
- Assembly of small nuclear ribonucleoproteins (snRNPs), essential components of the spliceosome that processes pre-messenger RNA
- Axonal transport in neurons
- Neuromuscular junction formation and maintenance
- Possibly other neuron-specific functions still being elucidated
Disease Modifier Genes: While SMN1 mutations are the primary cause, the clinical severity is strongly influenced by other genetic factors:
- SMN2 Gene: The most important disease modifier is the SMN2 gene, a paralog of SMN1 located in the same chromosomal region. Key aspects include:
- SMN2 is nearly identical to SMN1 but contains a critical C→T substitution in exon 7
- This substitution causes exon 7 to be excluded from most SMN2 transcripts (about 90%)
- The resulting protein (SMNΔ7) is unstable and rapidly degraded
- However, about 10-15% of SMN2 transcripts include exon 7 and produce full-length, functional SMN protein
- The number of SMN2 copies varies between individuals (typically 1-6 copies)
- Higher SMN2 copy numbers generally correlate with milder disease phenotypes
- Other Genetic Modifiers:
- Plastin 3 (PLS3): Higher expression has been associated with reduced symptom severity in females
- Neurocalcin delta (NCALD): Reduced levels may have protective effects
- ZPR1: Interacts with SMN and may modify disease severity
- NAIP (Neuronal Apoptosis Inhibitory Protein): Located in the same genomic region, deletions may contribute to severity
Cellular and Molecular Pathophysiology: The reduction in SMN protein leads to selective motor neuron degeneration through several mechanisms:
- Disrupted RNA Processing: Reduced SMN impairs snRNP assembly, affecting splicing of genes particularly important for motor neuron function
- Axonal Transport Defects: SMN deficiency disrupts the transport of mRNAs and proteins along the long axons of motor neurons
- Neuromuscular Junction Abnormalities: Reduced SMN affects the formation and maintenance of neuromuscular junctions
- Motor Neuron Vulnerability: Motor neurons appear particularly vulnerable to SMN deficiency, though the exact reasons remain incompletely understood
- Developmental Component: Some evidence suggests a developmental window where motor neurons are particularly dependent on adequate SMN levels
Hereditary Factors
Inheritance Pattern: SMA follows an autosomal recessive inheritance pattern:
- Both parents must be carriers (heterozygous for the SMN1 mutation) for a child to have SMA
- When both parents are carriers, each pregnancy has:
- 25% chance of having a child with SMA
- 50% chance of having a child who is a carrier
- 25% chance of having a child with two normal SMN1 copies
Carrier Frequency:
- Approximately 1 in 40-50 individuals in the general population are carriers
- The carrier frequency varies by ethnicity:
- Caucasians: 1 in 35
- Asians: 1 in 53
- African Americans: 1 in 66
- Hispanics: 1 in 117
Genetic Complexities:
- Silent Carriers: Some individuals have two SMN1 copies on one chromosome and none on the other, making them carriers despite having two SMN1 copies in total
- De Novo Mutations: About 2% of SMA cases result from new mutations rather than inherited mutations
- Intragenic Mutations: While most SMA cases involve complete deletion of SMN1, about 5% result from other mutations within the gene
- SMN2 Copy Number Variation: The number of SMN2 copies is inherited and influences disease severity
Known Triggers or Exposure Risks
Unlike many neurodegenerative diseases, SMA is a primarily genetic condition without well-established environmental triggers. However, several factors may influence disease onset or progression:
Potential Exacerbating Factors:
- Respiratory Infections: Can trigger rapid deterioration, especially in more severe types
- Fasting or Metabolic Stress: May temporarily worsen symptoms
- Fever: Can exacerbate weakness in some patients
- Rapid Growth Periods: Adolescent growth spurts can accelerate scoliosis progression
- Pregnancy: May temporarily worsen symptoms in female patients due to increased physiological demands
Hypothesized Environmental Influences: Some research has investigated potential environmental factors that might influence disease manifestation in genetically susceptible individuals:
- Oxidative Stress: Some studies suggest oxidative stress may contribute to motor neuron degeneration
- Maternal Diet During Pregnancy: Limited evidence suggests maternal nutrition might influence disease onset in affected infants
- Exposure to Certain Toxins: Some animal models suggest certain neurotoxins might accelerate motor neuron loss in the presence of SMN deficiency
It’s important to note that while these environmental factors may modify disease course, they are not causative—the fundamental cause remains the genetic mutation in SMN1. Unlike some neurological conditions where environmental exposures play a major causative role, SMA is primarily determined by genetics, with the environment playing at most a modifying role.
The understanding of SMA’s causes has been instrumental in developing targeted therapies that address the fundamental genetic defect or its consequences. All three currently approved therapies for SMA work by either replacing the SMN1 gene (gene therapy), enhancing SMN2 exon 7 inclusion (antisense oligonucleotides or small molecules), or providing the missing SMN protein through alternative approaches.
5. Risk Factors
Primary Risk Factors
Genetic Factors:
Family History and Carrier Status:
- The primary risk factor for having a child with SMA is when both parents are carriers of an SMN1 mutation
- Having a sibling with SMA significantly increases the risk for subsequent pregnancies
- Being a carrier (having one mutated SMN1 copy) does not cause symptoms but creates risk for offspring if partner is also a carrier
Consanguinity:
- Marriages between blood relatives increase the risk of autosomal recessive conditions like SMA
- In populations with higher rates of consanguineous marriages, SMA may be more prevalent
Ethnic Background:
- While SMA affects all populations, carrier frequencies vary by ethnicity:
- Higher carrier rates in Caucasian and Asian populations
- Relatively lower carrier rates in Hispanic and African populations
- Certain populations have founder mutations or higher rates of silent carriers
- While SMA affects all populations, carrier frequencies vary by ethnicity:
Geographic Ancestry:
- Some geographic regions show higher SMA prevalence due to founder effects or genetic isolation
- For example, higher carrier frequencies have been reported in parts of:
- Northern Europe
- Mediterranean region
- Parts of Asia
Demographic Risk Considerations
Age Factors:
- Parental Age: Unlike some genetic conditions, advanced parental age is not a significant risk factor for SMA
- Age of Onset: Age-related risk for clinical manifestation varies by SMA type:
- Type 0: Prenatal onset
- Type I: Highest risk in first 6 months of life
- Type II: Highest risk between 6-18 months
- Type III: Typically presents after 18 months, with variable age of onset
- Type IV: Onset in adulthood (typically 30s)
Gender Considerations:
- SMA affects males and females equally, consistent with its autosomal recessive inheritance
- Some emerging research suggests subtle gender differences in disease manifestation:
- Females with certain genotypes may have slightly milder phenotypes in some cases
- Potential protective effect of plastin 3 (PLS3) in some female carriers
- These differences are generally subtle and not clinically significant for risk assessment
Geographical and Environmental Factors:
- No strong evidence for environmental factors increasing SMA risk
- Geographic variations in prevalence primarily reflect:
- Differences in carrier frequencies
- Variability in diagnostic capabilities and reporting
- Differing rates of consanguinity
- Access to genetic counseling and prenatal testing
Special Risk Considerations
Genetic Testing Complexities:
- Silent Carriers: Some individuals have two SMN1 copies on one chromosome and none on the other. These “silent carriers” have a normal SMN1 copy count but can still pass the disease to offspring.
- Limitations of Testing: Standard carrier testing misses approximately 5% of carriers due to:
- Small intragenic mutations rather than whole gene deletions
- Silent carrier status
- De novo mutations
Risk Quantification:
- Carrier Couple: When both parents are carriers, each pregnancy has a 25% risk of SMA
- Known Carrier with Unknown Partner Status: Risk approximately 1 in 150-200 (variable by ethnicity)
- General Population Risk: Approximately 1 in 6,000-10,000 births
- Risk After Affected Child: 25% for each subsequent pregnancy (unless due to de novo mutation)
Impact of SMN2 Copy Number on Risk Classification:
- SMN2 copy number significantly influences disease severity but does not affect the risk of having the condition
- In prenatal diagnosis, SMN2 copy number analysis can provide prognostic information:
- 1-2 copies: Higher risk of Type I or II
- 3 copies: Often associated with Type II or III
- 4+ copies: Generally associated with milder phenotypes (Type III or IV)
- This information is valuable for prognostication but has limitations in precisely predicting disease course
Pre-existing Conditions: Unlike many diseases, pre-existing medical conditions generally do not increase the risk of developing SMA, as the condition is genetically determined. However:
Reproductive Technologies: Use of assisted reproductive technologies does not increase SMA risk, but genetic testing is recommended for donors in these contexts
Prenatal and Perinatal Factors: While not affecting the risk of having the genetic condition, some factors may influence disease onset or severity:
- Premature birth may complicate management of already-affected infants
- Birth trauma theoretically might accelerate symptom onset in predisposed infants
Neurological Comorbidities: Other neuromuscular conditions might complicate diagnosis or management but do not increase SMA risk
Understanding these risk factors is crucial for:
- Appropriate genetic counseling
- Identifying high-risk populations for carrier screening
- Guiding reproductive decision-making
- Informing newborn screening programs
- Early diagnosis and intervention
The landscape of risk assessment for SMA has evolved significantly with advances in genetic testing, increased availability of carrier screening, implementation of newborn screening, and the advent of effective therapies that can dramatically alter the natural history of the disease if initiated early.
6. Complications
Physical Complications
Respiratory Complications:
- Respiratory Insufficiency: Progressive weakness of intercostal muscles and diaphragm leads to:
- Impaired cough and secretion clearance
- Hypoventilation, particularly during sleep
- Recurrent respiratory infections
- Atelectasis (collapsed lung tissue)
- Respiratory Failure: The leading cause of mortality in untreated SMA, particularly Types I and II
- Sleep-Disordered Breathing: Including sleep apnea and nocturnal hypoventilation
- Chronic Respiratory Insufficiency: May eventually require non-invasive or invasive ventilatory support
- Pulmonary Function Decline: Progressive reduction in forced vital capacity and other measures
- Recurrent Pneumonia: Due to aspiration and inability to clear secretions
Musculoskeletal Complications:
- Scoliosis: Affects up to 80% of patients with Types II and III
- Progressive curvature due to paravertebral muscle weakness
- Often requires surgical intervention
- Can further compromise respiratory function
- Joint Contractures: Fixed limitations in joint movement due to muscle imbalance and disuse
- Particularly affects hips, knees, ankles, elbows, and wrists
- Can significantly impact function and care
- Hip Dysplasia and Dislocation: Due to muscle weakness and abnormal joint forces
- Osteopenia and Osteoporosis: Resulting from immobility, nutritional factors, and possibly intrinsic bone effects of SMN deficiency
- Fractures: Increased risk due to reduced bone density and falls
- Chest Wall Deformities: Including pectus excavatum and bell-shaped chest
Nutritional and Gastrointestinal Complications:
- Feeding Difficulties: Due to bulbar dysfunction and respiratory complications
- Weak suck in infants
- Difficulty swallowing and chewing
- Prolonged feeding times
- Fatigue during meals
- Dysphagia: Swallowing dysfunction increasing aspiration risk
- Gastroesophageal Reflux Disease (GERD): Common and potentially severe
- Constipation: Due to immobility, weak abdominal muscles, and altered gut motility
- Malnutrition: Can accelerate muscle loss and weaken respiratory function
- Growth Failure: Particularly in more severe types
Systemic Complications
Cardiovascular Complications:
- Cardiac Defects: Rare but reported in very severe SMA (Type 0/I), including:
- Atrial and ventricular septal defects
- Hypoplastic left heart
- Autonomic Dysfunction: May affect heart rate and blood pressure regulation
- Potential Cardiomyopathy: Some evidence for cardiac involvement, particularly in severe, untreated cases
- Peripheral Vascular Issues: Reduced circulation in limbs due to immobility
Metabolic Complications:
- Altered Energy Metabolism: Evidence for:
- Abnormal fatty acid metabolism
- Mitochondrial dysfunction
- Potential insulin resistance
- Growth Hormone Abnormalities: Some studies suggest altered growth hormone regulation
- Metabolic Acidosis: Can occur during illness or fasting
- Altered Body Composition: Reduced muscle mass and increased fat percentage
Neurological Complications:
- Motor Neuron Loss: Progressive degeneration of anterior horn cells
- Sensory Preservation: Sensory function typically unaffected
- Autonomic Involvement: In severe cases, may affect:
- Temperature regulation
- Sweating
- Gastrointestinal function
- Cardiovascular regulation
- Rare Central Nervous System Manifestations: In very severe cases (primarily Type 0)
Psychosocial Complications
Psychological Impact:
- Anxiety and Depression: Higher prevalence than in the general population
- Adjustment Disorders: Related to progressive disability
- Stress: Associated with medical procedures and hospitalizations
- Body Image Concerns: Particularly during adolescence
- Existential Distress: Confronting progressive condition and mortality
Social Challenges:
- Educational Barriers: Despite normal cognitive ability
- Employment Challenges: Workplace accessibility and accommodation issues
- Relationship Difficulties: Dating and partnership challenges
- Social Isolation: Due to mobility limitations, fatigue, and accessibility barriers
- Caregiver Burden: Significant impact on family caregivers
- Financial strain
- Physical demands
- Emotional toll
- Time requirements
Developmental and Quality of Life Impact:
- Delayed Independence: May affect developmental milestones and transitions
- Limited Participation: In age-appropriate activities
- Adaptations Required: For education, recreation, and daily living
- Pain and Fatigue: Affect quality of life and participation
- Sleep Disturbances: Common and impact daily functioning
Long-term Impact and Mortality
Long-term Organ System Impact:
- Progressive Muscle Atrophy: Continues throughout life, though rate varies by type
- Respiratory System Deterioration: Often determines prognosis in Types I and II
- Skeletal System Deformities: Progressive and often require surgical management
- Potential Subclinical Involvement: Of cardiac and other systems becoming apparent with longer survival
Disability Progression:
- Type I (Untreated):
- Typically fatal before age 2 years without ventilatory support
- Respiratory failure is the most common cause of death
- Type I (Treated with Current Therapies):
- Dramatically altered natural history
- Many achieving motor milestones previously thought impossible
- Long-term outcomes still being determined
- Type II:
- Variable lifespan, many reaching adulthood with appropriate support
- Progressive scoliosis and respiratory insufficiency
- Most require wheelchair mobility
- Type III:
- Near-normal lifespan
- About 50% lose independent ambulation by adolescence
- Progressive functional limitations but generally preserved respiratory function
- Type IV:
- Normal lifespan
- Slowly progressive weakness
- Typically maintains independence with adaptive equipment
Survival Statistics (Pre-Treatment Era):
- Type I: Median survival 8-10 months without ventilation
- Type II: Survival often into adulthood but reduced life expectancy
- Type III and IV: Near-normal life expectancy
Survival Statistics (Treatment Era):
- Emerging data shows dramatically improved survival across all types
- Type I patients receiving early treatment showing unprecedented motor function
- Long-term data still being collected, but trend shows:
- Reduced mortality
- Improved functional outcomes
- Reduced complication rates
- Shift in natural history of the disease
The landscape of SMA complications is changing dramatically with new treatments, early diagnosis through newborn screening, and improved supportive care. While the long-term impact of these advances is still being documented, they have already transformed the prognosis and management approach for many patients with SMA.
7. Diagnosis & Testing
Clinical Evaluation and Initial Assessment
Key Clinical Features Prompting Diagnosis:
- Hypotonia (low muscle tone/”floppiness”)
- Proximal muscle weakness (shoulders, hips, trunk)
- Absent or diminished deep tendon reflexes
- Preservation of sensation and cognitive function
- Symmetric muscle involvement
- Fasciculations, particularly of the tongue (in infantile forms)
- Normal or enhanced cognitive development
Initial Clinical Assessment:
- Detailed History:
- Motor milestone achievement or regression
- Family history of neuromuscular disorders
- Pregnancy and birth history
- Pattern and progression of weakness
- Physical Examination:
- Neurological assessment focusing on motor function
- Musculoskeletal evaluation
- Respiratory assessment
- Evaluation of feeding/swallowing
- Documentation of contractures or deformities
- Functional Assessments:
- Age-appropriate motor function tests
- Standardized scales (CHOP INTEND, HFMSE, RULM, 6MWT)
- Respiratory function testing (when applicable)
Clinical Red Flags for SMA:
- Proximal weakness greater than distal
- “Frog-leg” posture in infants
- Absent reflexes with preserved sensation
- Progressive course
- Alert, engaged appearance despite significant weakness
- Fasciculations (especially of the tongue)
- Bell-shaped chest or paradoxical breathing
Diagnostic Laboratory Testing
Genetic Testing (Gold Standard):
- SMN1 Deletion/Mutation Analysis:
- PCR and MLPA (Multiplex Ligation-dependent Probe Amplification) to detect homozygous deletion of SMN1 exon 7
- Identifies approximately 95% of SMA cases
- First-line diagnostic test when SMA is suspected
- SMN1 Sequencing:
- For patients with clinical SMA but negative deletion testing
- Identifies small intragenic mutations
- Detects approximately 5% of cases (those without deletions)
- SMN2 Copy Number Analysis:
- Not diagnostic but provides important prognostic information
- Higher copy numbers generally correlate with milder phenotypes
- Increasingly important for treatment decisions and clinical trial eligibility
Supportive Laboratory Tests:
- Creatine Kinase (CK):
- Normal or mildly elevated (typically less than 10x normal)
- Contrasts with much higher elevations in muscular dystrophies
- Liver Function Tests:
- May show mild transaminase elevations despite normal liver function
- Related to muscle involvement rather than liver disease
- Complete Blood Count and Metabolic Panel:
- Generally normal but important to rule out other conditions
- May show signs of metabolic stress during acute illness
Electrophysiological Studies:
- Electromyography (EMG):
- Shows denervation potentials
- Fibrillations and positive sharp waves
- Fasciculations
- Large motor unit potentials
- Nerve Conduction Studies (NCS):
- Normal sensory nerve conduction
- Reduced compound muscle action potentials
- Normal or mildly reduced conduction velocities
- While historically important, EMG/NCS are now often deferred in favor of genetic testing, particularly in infants
Specialized Testing and Imaging
Muscle Biopsy:
- Rarely performed now that genetic testing is widely available
- Historical findings include:
- Grouped atrophy (groups of atrophic fibers adjacent to normal or hypertrophic fibers)
- Type grouping (evidence of denervation and reinnervation)
- No primary muscle pathology
- Both fiber types affected
Imaging Studies:
- Spine Radiographs:
- Assessment of scoliosis and pelvic obliquity
- Monitoring of progression
- Surgical planning
- Musculoskeletal Ultrasound:
- Evaluation of muscle architecture
- Monitoring of muscle thickness and echogenicity
- Non-invasive and increasingly used in research
- Magnetic Resonance Imaging (MRI):
- Spinal MRI to assess cord integrity and rule out structural lesions
- Muscle MRI showing characteristic patterns of fatty replacement
- Research tool for quantifying muscle replacement
- Dual-Energy X-ray Absorptiometry (DEXA):
- Assessment of bone mineral density
- Evaluation of fracture risk
Respiratory Assessment:
- Pulmonary Function Testing (when applicable):
- Spirometry with forced vital capacity (FVC)
- Maximum inspiratory and expiratory pressures
- Overnight Sleep Studies:
- Detection of sleep-disordered breathing
- Assessment of nocturnal hypoventilation
- End-tidal or transcutaneous CO2 monitoring:
- Evaluation for hypercapnia
- Cough peak flow measurement:
- Assessment of cough effectiveness
Nutritional and Swallowing Assessment:
- Videofluoroscopic Swallow Study:
- Evaluation of swallowing safety and aspiration risk
- Fiberoptic Endoscopic Evaluation of Swallowing (FEES):
- Direct visualization of swallowing function
- Nutritional laboratory parameters:
- Albumin, prealbumin, vitamins, etc.
Early Detection and Screening
Newborn Screening:
- Increasingly implemented internationally
- Methods include:
- Initial screening for low SMN1 exon 7 levels
- Second-tier testing for SMN2 copy number
- Confirmation with full genetic analysis
- Benefits:
- Allows pre-symptomatic diagnosis
- Enables treatment before motor neuron loss
- Dramatically improves outcomes, especially for Type I
- Limitations:
- May identify patients with 4+ SMN2 copies who might develop very mild disease
- Creates medical and ethical challenges regarding when to treat
- Access to follow-up and treatment varies globally
Carrier Screening:
- Preconception Carrier Screening:
- Increasingly offered as part of expanded carrier panels
- Can identify couples at risk before pregnancy
- Prenatal Carrier Screening:
- Testing of parents during pregnancy
- Enables informed reproductive decisions
- Limitations:
- Does not detect all carriers (misses ~5%)
- Does not identify SMN2 copy number in carriers
- Variable implementation and access globally
Prenatal Diagnosis:
- Methods:
- Chorionic villus sampling (11-13 weeks)
- Amniocentesis (15-20 weeks)
- Non-invasive prenatal testing (emerging techniques, not yet widely available)
- Approaches:
- Direct testing for SMN1 deletions/mutations
- SMN2 copy number analysis for prognostication
- Linkage analysis when familial mutation is unknown
Pre-implantation Genetic Diagnosis (PGD):
- Used with in vitro fertilization
- Testing of embryos for SMN1 mutations
- Selection of unaffected embryos for implantation
- Can include SMN2 copy number analysis
Diagnostic Challenges and Differential Diagnosis
Diagnostic Challenges:
- Mild Phenotypes: May have delayed diagnosis, particularly Type IV
- Prenatal/Neonatal Onset: May be confused with other congenital disorders
- Atypical Presentations: Cases with preserved reflexes or asymmetrical features
- Complex Genotypes: Difficult interpretation with certain SMN1/SMN2 configurations
- Limited Testing Access: In resource-limited settings
Differential Diagnosis:
- Other Lower Motor Neuron Diseases:
- Hereditary motor neuropathies
- Infantile neuronal degeneration
- X-linked infantile spinal muscular atrophy
- Congenital Myopathies:
- Nemaline myopathy
- Centronuclear myopathy
- Congenital fiber-type disproportion
- Muscular Dystrophies:
- Congenital muscular dystrophies
- Limb-girdle muscular dystrophies
- Myotonic dystrophy
- Metabolic and Mitochondrial Disorders:
- Pompe disease
- Fatty acid oxidation disorders
- Mitochondrial myopathies
- Neuromuscular Junction Disorders:
- Congenital myasthenic syndromes
- Infantile botulism
- Non-neuromuscular Conditions:
- Prader-Willi syndrome
- Hypotonic cerebral palsy
- Congenital hypotonia due to brain injury
The diagnostic approach to SMA has evolved dramatically with advances in genetic testing. Modern diagnosis typically relies on genetic confirmation, with supportive clinical and electrophysiological data. The implementation of newborn screening has enabled pre-symptomatic diagnosis, fundamentally changing the therapeutic approach and improving outcomes. However, diagnostic challenges remain, particularly in resource-limited settings and for patients with atypical presentations or complex genotypes.
8. Treatment Options
Disease-Modifying Therapies
The treatment landscape for SMA has been transformed by the approval of three disease-modifying therapies, each with a different mechanism of action but all targeting the underlying genetic defect:
Nusinersen (Spinraza):
- Mechanism: Antisense oligonucleotide that modifies SMN2 pre-mRNA splicing to increase exon 7 inclusion and full-length SMN protein production
- Administration: Intrathecal injection (via lumbar puncture)
- Dosing Schedule:
- Loading doses at days 0, 14, 28, and 63
- Maintenance doses every 4 months thereafter
- FDA Approval: December 2016
- Approved Population: All SMA types and ages
- Efficacy Data:
- ENDEAR trial (infantile-onset): 51% responders in motor milestone development vs. 0% in control group
- CHERISH trial (later-onset): Significant improvement in motor function scores
- Long-term follow-up studies showing sustained benefit for most patients
- Considerations:
- Requires specialized administration
- Potential procedural complications
- Challenging in patients with severe scoliosis
- Lifelong treatment required
- High cost (approximately $125,000-$150,000 per dose)
Onasemnogene Abeparvovec-xioi (Zolgensma):
- Mechanism: Gene replacement therapy using adeno-associated virus (AAV9) vector to deliver functional copy of human SMN1 gene
- Administration: One-time intravenous infusion
- FDA Approval: May 2019
- Approved Population: Children under 2 years with SMA (US); varied age/weight limits internationally
- Efficacy Data:
- STR1VE trial: 91% of patients achieved event-free survival at 14 months
- Significant improvement in motor milestone achievement
- Sustained expression of SMN protein years after treatment
- Considerations:
- One-time treatment
- Extremely high cost ($2.1 million per treatment)
- Pre-existing AAV9 antibodies may exclude some patients
- Requires liver function monitoring
- Potential for immune response to the vector
- Limited long-term safety and efficacy data beyond 5-7 years
- Some patients may require other therapies later in life
Risdiplam (Evrysdi):
- Mechanism: Small molecule that modifies SMN2 splicing to increase full-length SMN protein production
- Administration: Daily oral liquid
- FDA Approval: August 2020
- Approved Population: Patients 2 months of age and older with all SMA types
- Efficacy Data:
- FIREFISH trial (Type I): 41% sitting without support after 12 months
- SUNFISH trial (Types II/III): Significant improvement in motor function
- Convenience of oral administration with good CNS penetration
- Considerations:
- First oral therapy for SMA
- Weight-based dosing
- Requires regular dosing schedule
- Potential drug interactions
- Ongoing assessment of long-term outcomes
- Annual cost approximately $340,000 (varies by weight)
Combination Approaches:
- Emerging evidence for potential benefits of combination therapy
- Various approaches under investigation:
- Sequential therapy (typically gene therapy followed by SMN2-modifying treatment)
- Concurrent therapy with complementary mechanisms
- Safety and efficacy data still emerging
- No formal guidelines yet established
Supportive Care and Management
Respiratory Management:
- Airway Clearance Techniques:
- Manual and mechanical cough assistance
- Chest physiotherapy
- Postural drainage
- Ventilatory Support:
- Non-invasive ventilation (NIV) – typically bilevel positive airway pressure (BiPAP)
- Invasive ventilation via tracheostomy when necessary
- Mechanically assisted cough devices
- Respiratory Monitoring:
- Regular pulmonary function testing when applicable
- Sleep studies
- End-tidal or transcutaneous CO2 monitoring
- Immunizations:
- Influenza vaccine
- Pneumococcal vaccines
- Respiratory syncytial virus (RSV) prophylaxis in infants
Nutritional and Gastrointestinal Management:
- Feeding Approaches:
- Specialized feeding techniques for oral feeding
- Positioning adjustments
- Modified food textures
- Supplemental tube feeding when necessary
- Nutritional Support:
- Optimized caloric intake
- Appropriate macronutrient distribution
- Vitamin and mineral supplementation
- Management of Complications:
- Gastroesophageal reflux treatment
- Constipation management
- Monitoring for aspiration
Orthopedic and Rehabilitation Management:
- Physical Therapy:
- Range of motion exercises
- Strengthening within capability
- Positioning and postural support
- Aquatic therapy
- Occupational Therapy:
- Activities of daily living adaptation
- Upper extremity function optimization
- Assistive device training
- Orthotics and Bracing:
- Ankle-foot orthoses (AFOs)
- Thoracolumbosacral orthoses (TLSOs) for scoliosis
- Custom seating systems
- Surgical Interventions:
- Scoliosis correction
- Hip subluxation/dislocation management
- Contracture release procedures
Assistive Technology and Mobility:
- Mobility Aids:
- Manual wheelchairs (often power-assist)
- Power wheelchairs with specialized controls
- Standing frames and gait trainers
- Communication Devices:
- Augmentative and alternative communication (AAC)
- Environmental control systems
- Computer access adaptations
- Home Modifications:
- Accessibility adaptations
- Lift systems
- Adaptive furniture
Psychosocial Support:
- Psychological Services:
- Coping strategies
- Management of anxiety and depression
- Family therapy
- Educational Support:
- Individualized education plans
- School accommodations
- Vocational planning for older patients
- Family Resources:
- Caregiver training
- Respite care
- Support groups
- Financial resource guidance
Emerging Treatments and Clinical Trials
Advanced SMN-Based Approaches:
- Novel SMN2 Splicing Modifiers:
- Next-generation splice modulators with enhanced potency
- Extended-release formulations
- Tissue-targeted delivery systems
- SMN Protein Stabilizers:
- Small molecules that prevent SMN protein degradation
- Compounds that enhance SMN protein function
- Alternative Gene Therapy Approaches:
- Next-generation viral vectors
- Non-viral gene delivery systems
- Regulatable gene expression systems
Non-SMN Based Approaches:
- Neuroprotective Agents:
- Compounds that protect motor neurons independent of SMN
- Anti-apoptotic agents
- Neurotrophic factors
- Muscle-Enhancing Therapies:
- Myostatin inhibitors
- Troponin activators
- Muscle stem cell approaches
- Neuromuscular Junction (NMJ) Enhancers:
- Compounds that improve NMJ function
- Acetylcholine receptor modulators
- NMJ maintenance factors
Selected Notable Clinical Trials:
- Branaplam (LMI070):
- Oral SMN2 splicing modifier
- Development suspended due to safety signals
- SRK-015/Apitegromab:
- Myostatin inhibitor as add-on therapy
- Phase 2 TOPAZ trial showed some functional benefits
- Reldesemtiv:
- Fast skeletal muscle troponin activator
- Aims to increase muscle strength directly
- ION363/Jacifusen:
- Antisense oligonucleotide targeting FUS for ALS, being explored in SMA
- Combination Therapy Trials:
- Various studies evaluating sequential or concurrent treatment with approved therapies
Challenges in Treatment Landscape:
- Access and Cost Issues:
- Extremely high therapy costs
- Insurance coverage barriers
- Global inequities in access
- Resource allocation ethical challenges
- Timing of Intervention:
- Pre-symptomatic vs. symptomatic treatment
- Optimization of treatment timing
- Selection criteria for different therapies
- Long-Term Outcomes:
- Durability of treatment effects
- Potential late-appearing adverse effects
- Impact on non-motor aspects of SMA
- Biomarker Development:
- Need for better predictive and response biomarkers
- Identification of patients most likely to benefit from specific treatments
- Monitoring tools for long-term follow-up
Future Treatment Paradigms:
- Increasing personalization based on:
- Age at diagnosis
- SMA type and severity
- Functional status
- SMN2 copy number
- Prior treatments
- Patient/family preferences
- Moving toward combination approaches targeting multiple aspects of the disease
- Integration of technology and therapy
- Comprehensive care models incorporating disease-modifying and supportive therapies
The treatment landscape for SMA has been revolutionized in recent years, transitioning from purely supportive care to disease-modifying therapies that can dramatically alter the disease course. Early diagnosis and treatment, particularly through newborn screening, offers the best outcomes, though significant benefit can be seen even in later-onset cases. While current approved therapies primarily address the genetic cause, emerging approaches target multiple mechanisms, potentially offering additional benefits and addressing aspects of the disease not fully managed by SMN restoration alone.
9. Prevention & Precautionary Measures
Genetic Counseling and Family Planning
Carrier Screening:
- General Population Screening:
- Increasingly included in expanded carrier screening panels
- Recommended for consideration in all couples planning pregnancy
- Particularly important in populations with higher carrier frequency
- Targeted Screening:
- Recommended for family members of SMA patients
- Partners of known carriers
- Couples with family history of undiagnosed neuromuscular disorders
- Testing Approach:
- Quantitative analysis of SMN1 copy number
- Cannot identify all carriers (misses “2+0” carriers with two copies on one chromosome)
- Approximately 95% detection rate for carriers
Prenatal Testing Options:
- Invasive Prenatal Diagnosis:
- Chorionic Villus Sampling (CVS): Performed at 10-13 weeks gestation
- Amniocentesis: Performed at 15-20 weeks gestation
- Both test for SMN1 deletion/mutation and can assess SMN2 copy number
- Near 100% accuracy for diagnosis
- Small procedural risk of pregnancy loss (0.1-0.5%)
- Non-Invasive Prenatal Testing (NIPT):
- Emerging techniques using cell-free fetal DNA
- Not yet widely available for SMA
- Research ongoing to improve accuracy and availability
- Preimplantation Genetic Testing (PGT):
- Used with in vitro fertilization (IVF)
- Embryo biopsy and genetic analysis before implantation
- Selection of embryos without SMA-causing mutations
- Can include SMN2 copy number analysis
- Complex and costly process but avoids termination decisions
Reproductive Options for At-Risk Couples:
- Natural conception with prenatal testing
- Preimplantation genetic testing with IVF
- Use of donor gametes (egg or sperm)
- Adoption
- Decision to accept the risk without intervention
Psychological Aspects of Genetic Counseling:
- Coping with carrier status
- Decision-making support around reproductive options
- Guilt and family dynamics
- Cultural and religious considerations
- Ethical discussions around testing and prevention
Newborn Screening and Early Intervention
Newborn Screening Programs:
- Implementation Status:
- Increasingly adopted in many countries and regions
- Added to Recommended Uniform Screening Panel (RUSP) in the US in 2018
- Varied implementation internationally
- Screening Methods:
- First-tier: Quantitative PCR or MLPA to detect SMN1 exon 7 deletion
- Second-tier: SMN2 copy number analysis for positive screens
- Confirmatory testing for definitive diagnosis
- Benefits of Newborn Screening:
- Pre-symptomatic diagnosis
- Treatment initiation before significant motor neuron loss
- Dramatically improved outcomes, especially for Type I SMA
- Family planning information for future pregnancies
- Challenges and Considerations:
- Detection of patients with 4+ SMN2 copies who might develop very mild disease
- Medical and ethical challenges regarding when to treat
- Access to follow-up and treatment varies globally
- Cost-effectiveness depends on treatment access
Early Intervention Protocols:
- Immediate Referral Pathways:
- Rapid referral to specialized centers
- Comprehensive baseline evaluations
- Initiation of treatment as indicated
- Pre-symptomatic Treatment:
- Decisions based on SMN2 copy number and family history
- Treatment protocols for pre-symptomatic patients
- Monitoring for development of symptoms
- Multidisciplinary Follow-up:
- Regular neurological assessments
- Standardized functional assessments
- Proactive respiratory monitoring
- Nutritional support
- Early rehabilitation interventions
Preventive Care for Diagnosed Patients
While there is no way to prevent SMA after diagnosis, several measures can prevent or minimize complications:
Respiratory Preventive Measures:
- Immunizations:
- Annual influenza vaccine
- Pneumococcal vaccines
- COVID-19 vaccination
- RSV prophylaxis in infants when indicated
- Proactive Respiratory Management:
- Regular pulmonary function monitoring
- Early introduction of cough assistance
- Preventive chest physiotherapy
- Sleep studies to detect early hypoventilation
- Avoiding respiratory irritants and exposures
- Infection Prevention:
- Hand hygiene protocols
- Limiting exposure during epidemic seasons
- Prompt treatment of respiratory infections
- Education on early warning signs
Musculoskeletal Preventive Measures:
- Contracture Prevention:
- Regular stretching programs
- Proper positioning
- Orthotic use
- Range of motion exercises
- Scoliosis Monitoring and Management:
- Regular spine examinations
- Radiographic monitoring
- Early bracing when indicated
- Proactive surgical intervention when necessary
- Osteoporosis Prevention:
- Adequate calcium and vitamin D
- Weight-bearing activities when possible
- Consideration of bone density monitoring
- Fall prevention strategies
Nutritional Preventive Strategies:
- Aspiration Prevention:
- Proper feeding techniques
- Appropriate food textures
- Positioning during and after meals
- Swallowing evaluations
- Optimized Nutrition:
- Balanced nutritional intake
- Appropriate caloric provision
- Monitoring for undernutrition or overnutrition
- Early intervention for feeding difficulties
- Gastrointestinal Complication Prevention:
- Prophylactic management of constipation
- Proactive GERD prevention and treatment
- Hydration protocols
Preventive Health Maintenance:
- Regular Health Surveillance:
- Primary care coordination
- Dental hygiene and care
- Age-appropriate health screenings
- Symptom monitoring protocols
- Psychological Support:
- Regular assessment of psychological well-being
- Early intervention for anxiety or depression
- Family support systems
- Coping strategy development
- Educational and Developmental Support:
- Early intervention services
- Educational accommodations
- Vocational planning
- Life transitions preparation
Environmental and Lifestyle Considerations
While SMA is a genetic condition not caused by environmental factors, certain environmental and lifestyle considerations may influence quality of life and disease management:
Environmental Modifications:
- Home Adaptations:
- Accessibility modifications
- Assistive technology implementation
- Temperature control (temperature sensitivity is common)
- Air quality optimization (HEPA filters, humidity control)
- School/Workplace Accommodations:
- Physical accessibility
- Adapted equipment
- Modified schedules to manage fatigue
- Emergency protocols
Activity Management:
- Energy Conservation:
- Pacing strategies
- Prioritization of activities
- Scheduled rest periods
- Adaptive techniques
- Appropriate Exercise:
- Individualized exercise plans
- Aquatic therapy
- Non-fatiguing strengthening
- Recreational adaptations
- Participation Optimization:
- Adaptive sports and recreation
- Modified participation strategies
- Technology utilization
- Peer connection opportunities
Exposure Prevention:
- Infection Control:
- Enhanced hygiene practices
- Avoiding high-risk exposure situations
- Early isolation during illness outbreaks
- Visitor screening during vulnerable periods
- Extreme Weather Precautions:
- Temperature regulation strategies
- Emergency power plans for equipment
- Evacuation protocols
- Heat and cold stress prevention
Long-term Planning:
- Transition Planning:
- Pediatric to adult care transitions
- Educational transitions
- Independent living preparation
- Financial planning
- Advance Care Planning:
- Discussion of treatment preferences
- Emergency care plans
- Documentation of wishes
- Surrogate decision-maker designation
The prevention landscape for SMA has evolved dramatically with advances in genetic testing, newborn screening, and treatment options. While primary prevention through genetic counseling remains important, the focus has increasingly shifted to pre-symptomatic detection and early intervention, which can dramatically alter disease course. For diagnosed patients, prevention of complications through proactive, multidisciplinary care is essential to maximize function and quality of life.
10. Global & Regional Statistics
Global Prevalence and Incidence
Worldwide Prevalence:
- Overall Prevalence: Approximately 1-2 per 100,000 individuals globally
- Incidence: Approximately 1 in 6,000 to 1 in 10,000 live births
- Total Cases: Estimated 300,000-400,000 individuals living with SMA worldwide
- Type Distribution:
- Type I: 60% of cases
- Type II: 27% of cases
- Type III: 12% of cases
- Type IV: <1% of cases
- Gender Distribution: Affects males and females equally (autosomal recessive inheritance)
Carrier Frequency:
- Average Global Carrier Frequency: Approximately 1 in 40-50 individuals
- Ethnic Variations in Carrier Rates:
- Caucasians: 1 in 35-40
- Asians: 1 in 40-60
- African Americans: 1 in 60-70
- Hispanics: 1 in 50-60
- Ashkenazi Jews: 1 in 41
- SMN1 Deletion Types by Population:
- Single-copy SMN1 deletions most common globally
- Higher rates of gene conversion events in certain Asian populations
- Variable rates of small intragenic mutations
Age Distribution:
- Age of Onset by Type:
- Type 0: Prenatal
- Type I: 0-6 months
- Type II: 6-18 months
- Type III: >18 months
- Type IV: Adult onset (>21 years)
- Age Distribution of Living Patients:
- Historically skewed toward Types II and III due to higher mortality in Type I
- Changing demographics with new treatments increasing Type I survival
- Growing adult population with SMA due to improved care and outcomes
Regional Statistics and Variations
North America:
- United States:
- Estimated 10,000-25,000 individuals living with SMA
- Carrier frequency approximately 1 in 40-54
- Comprehensive newborn screening implemented in most states
- High access to approved therapies
- Annual cost of care: $50,000-$250,000 (varies by type, excluding therapy costs)
- Canada:
- Estimated 2,500-3,000 individuals with SMA
- Variable provincial implementation of newborn screening
- Negotiated lower therapy costs through national review processes
- Comprehensive coverage through provincial health plans
Europe:
- Prevalence: 1-2 per 100,000 across Europe
- Screening Implementation: Variable by country
- Germany, Belgium, and Poland with established programs
- Many countries in planning or pilot phases
- Treatment Access: Varies significantly by country
- Western European countries with broader access
- Eastern and Southern European countries with more restricted access
- Notable Regional Initiatives:
- European Reference Network for Neuromuscular Diseases
- TREAT-NMD global registry
Asia:
- East Asia:
- Japan: Estimated 5,000 patients, increasing treatment access
- China: Approximately 30,000 patients, limited treatment access outside major cities
- South Korea: Comprehensive care centers developing rapidly
- South Asia:
- India: Large affected population but limited diagnosis and treatment
- Variable carrier frequencies reported across different ethnic groups
- Limited access to genetic testing and treatments
- Regional Characteristics:
- Higher prevalence of compound heterozygotes in some populations
- Emergence of regional care centers in major urban areas
- Growing treatment access but significant economic barriers
Middle East and Africa:
- Middle East:
- Higher SMA rates in areas with consanguineous marriage
- Emerging specialized care centers in Israel, Saudi Arabia, UAE
- Variable treatment access even within countries
- Africa:
- Limited epidemiological data
- Significant underdiagnosis likely
- Extremely limited access to genetic testing and treatments
- Few specialized care centers
- Growing awareness but substantial resource limitations
Australia and New Zealand:
- Well-established patient registries
- Estimate of 600-800 patients in Australia
- Comprehensive multidisciplinary care
- Growing implementation of newborn screening
- Universal healthcare systems facilitating treatment access
Latin America:
- Estimated 25,000 individuals living with SMA
- Variable care infrastructure by country
- Brazil and Mexico with largest patient populations
- Growing regional advocacy networks
- Limited but increasing treatment access
- Significant economic challenges for high-cost therapies
Mortality and Survival Data
Historical Survival Rates (Pre-Treatment Era):
- Type I (Severe):
- Median survival: 7-8 months without ventilation
- <10% survival beyond age 2 without ventilatory support
- Respiratory failure as primary cause of death
- Type II (Intermediate):
- Survival into adulthood common
- Median life expectancy: 20s-30s
- Progressive respiratory insufficiency as main cause of mortality
- Type III (Mild):
- Near-normal life expectancy
- Reduced life expectancy in some cases due to respiratory complications
- Type IV (Adult Onset):
- Normal life expectancy
- Minimal impact on mortality
Contemporary Survival Data (Treatment Era):
- Type I with Treatment:
- Dramatic improvement in survival
90% survival at 2 years in treated cohorts
- Long-term outcomes still being collected
- Event-free survival (no permanent ventilation) significantly improved
- All Types with Early Treatment:
- Emerging data shows significant alteration of natural history
- Achievement of motor milestones previously thought impossible
- Long-term outcome data still accumulating
- Shifting cause of death from early respiratory failure to later complications
Factors Affecting Survival:
- Age at Treatment Initiation:
- Earlier treatment strongly correlated with better outcomes
- Pre-symptomatic treatment shows best results
- Access to Multidisciplinary Care:
- Significant regional variations in survival based on care access
- Specialized center care associated with better outcomes
- Ventilation Decisions:
- Ventilatory support significantly extends survival
- Quality of life considerations impact ventilation choices
- Nutritional Management:
- Adequate nutrition strongly associated with better outcomes
- Proactive feeding interventions improve survival
- Treatment Type:
- Limited comparative data between approved therapies
- All show significant benefits compared to natural history
- Emerging data on combination approaches
Trends and Future Projections
Changing Epidemiology:
- Growing Prevalence: Increasing due to improved survival
- Altered Type Distribution: More surviving Type I patients
- Aging Population: Growing adult SMA population
- New Phenotypes: Treated patients showing distinct clinical patterns from natural history
- Treatment-Naïve vs. Treated Populations: Diverging natural histories
Healthcare Utilization Trends:
- Shifting Resource Needs:
- Decreased acute hospitalizations
- Increased outpatient and supportive care
- Changing orthopedic intervention patterns
- Evolving respiratory support requirements
- Economic Impact:
- High therapy costs ($100,000-$2.1 million)
- Reduced hospitalization costs
- Changing long-term care needs
- Growing economic impact studies
Regional Development Patterns:
- Expanding Newborn Screening:
- Rapid global expansion
- Resource-stratified implementation models emerging
- Treatment Access Expansion:
- Increasing global availability of approved therapies
- Emergence of alternative access programs
- Pricing challenges and innovation
- Care Center Development:
- Growth of specialized multidisciplinary centers
- Telemedicine models for remote populations
- Standardization of care protocols
Future Epidemiological Projections:
- Incidence: Expected to remain stable (genetic condition)
- Prevalence: Projected to increase due to improved survival
- Mortality: Continuing decline, particularly in early-onset types
- Diagnostic Timing: Shift toward newborn and pre-symptomatic diagnosis
- Global Disparities: Concern for widening gap between high and low-resource regions
- Long-term Outcomes: Emerging data will better define new natural history
The global landscape of SMA is evolving rapidly with advances in diagnosis, treatment, and care. While the genetic incidence remains stable, prevalence is increasing as patients live longer, creating new challenges for healthcare systems. Significant regional disparities persist in diagnosis and treatment access, with profound implications for outcomes. The next decade will likely see further improvements in survival balanced against the challenges of ensuring global equity in access to these advances.
11. Recent Research & Future Prospects
Latest Treatment Advancements
Refinement of Existing Therapies:
- Nusinersen (Spinraza) Advances:
- Alternative delivery methods under investigation:
- Subcutaneous formulations
- Intrathecal ports for easier administration
- Extended-interval dosing protocols
- Long-term outcome data showing sustained benefits
- Emerging data in previously understudied populations (adults, very young infants)
- Alternative delivery methods under investigation:
- Onasemnogene Abeparvovec (Zolgensma) Developments:
- Extended age and weight inclusion criteria
- Management protocols for side effects
- Strategies for patients with AAV9 antibodies
- Long-term durability data emerging
- Post-authorization safety monitoring
- Risdiplam (Evrysdi) Progress:
- Expanded age indications
- Simplified administration protocols
- Real-world effectiveness data
- Combination approaches with other therapies
Novel Therapeutic Approaches:
- SMN-Independent Therapies:
- SRK-015/Apitegromab: Myostatin inhibitor enhancing muscle function
- Reldesemtiv: Fast skeletal muscle troponin activator
- Oral HDAC Inhibitors: Targeting downstream pathways
- Anti-inflammatory Approaches: Addressing neuromuscular junction inflammation
- Enhanced Delivery Systems:
- Brain-penetrant small molecules
- Blood-brain barrier penetrating vectors
- Targeted delivery to specific cell types
- Controlled release formulations
- Second-Generation Gene Therapies:
- Regulatable gene expression systems
- Re-dosable gene therapy approaches
- Novel vectors with reduced immunogenicity
- Cell-specific targeting strategies
Combination Therapy Paradigms:
- Sequential Therapy Protocols:
- Gene therapy followed by SMN2-modifying agents
- SMN-enhancing therapy plus muscle-directed therapy
- Personalized combination approaches based on response
- Complementary Mechanism Combinations:
- SMN-dependent plus SMN-independent approaches
- Central plus peripheral acting agents
- Neuroprotective plus muscle-enhancing strategies
- Evidence Development:
- Emerging clinical data on combination safety
- Optimization of timing and sequencing
- Development of combination-specific endpoints
Ongoing Research Initiatives
Clinical Trial Landscape:
- Natural History Studies:
- Evolving natural history in the treatment era
- Long-term follow-up of treated patients
- Age and population-specific natural history
- Biomarker Development:
- Neurofilament light chain (NfL) as neurodegeneration marker
- Electrophysiological biomarkers
- Imaging biomarkers of muscle and nerve integrity
- Digital biomarkers from wearable technology
- Novel Outcome Measures:
- Development of sensitive, clinically meaningful endpoints
- Patient-reported outcome measures
- Application of technology in outcome assessment
- Predictive modeling of treatment response
Basic and Translational Research:
- Disease Mechanisms:
- Further understanding of SMN functions
- Non-neuronal contributions to pathophysiology
- Cellular and molecular mechanisms of motor neuron vulnerability
- Developmental versus degenerative components
- Animal Models:
- Refined models reflecting human disease
- Humanized models with patient-specific mutations
- Models for testing combination approaches
- Aged models for adult-onset disease
- Cellular Models:
- Patient-derived induced pluripotent stem cell models
- Organoid systems modeling neuromuscular development
- High-throughput screening platforms
- “Disease in a dish” for personalized medicine
Global Research Networks:
- Collaborative Research Platforms:
- TREAT-NMD global network
- European Reference Network for Neuromuscular Diseases
- SMA Newborn Screening Alliance
- International SMA Consortium
- Data Sharing Initiatives:
- Global patient registries
- Biobanking networks
- Real-world data collection frameworks
- Collaborative natural history databases
- Standardization Efforts:
- Harmonized outcome measures
- Standard operating procedures for assessments
- Cross-trial data comparability initiatives
- Common data elements for research
Future Therapeutic Directions
Emerging Therapeutic Targets:
- Alternative Splicing Modulators:
- Next-generation SMN2 splicing modifiers
- Targeted RNA editing approaches
- RNA trans-splicing strategies
- Protective Pathway Enhancement:
- STATHMIN-2 modulation
- Neuronal calcium regulation
- Mitochondrial protection strategies
- Axonal transport enhancement
- Neuromuscular Junction Optimization:
- Targeted enhancement of synaptic function
- Neurotrophic factor delivery
- Promotion of compensatory reinnervation
- Stabilization of existing connections
Advanced Therapeutic Modalities:
- Gene Editing Approaches:
- CRISPR/Cas9 correction of SMN1 mutations
- Base editing technologies
- Prime editing for precise genetic correction
- In vivo editing strategies
- RNA Therapeutics Beyond Splicing Modulation:
- mRNA delivery of SMN protein
- RNA interference targeting modifiers
- Circular RNA strategies
- Long non-coding RNA modulators
- Cell-Based Therapies:
- Motor neuron replacement strategies
- Supportive glial cell transplantation
- Genetically modified mesenchymal stem cells
- Induced neural progenitor approaches
Personalized Medicine Approaches:
- Genetic Modifier-Based Stratification:
- SMN2 copy number-guided therapy selection
- Additional modifier gene analysis
- Pharmacogenomic considerations
- Response-Based Treatment Algorithms:
- Adaptive treatment protocols
- Predictive modeling of optimal therapy
- Sequential therapy decision trees
- Biomarker-guided therapy adjustment
- Patient-Specific Considerations:
- Age-appropriate therapeutic approaches
- Accounting for disease duration and progression
- Comorbidity-adapted treatment plans
- Quality of life prioritization
Potential Cures and Transformative Therapies
Definitive Genetic Correction:
- In Vivo Gene Editing:
- Direct correction of SMN1 mutations
- Permanent genetic solution
- One-time curative potential
- Challenges in delivery and safety
- Advanced Gene Replacement:
- Next-generation vectors with improved safety
- Re-dosable approaches
- Regulated expression systems
- Cell type-specific delivery
Regenerative Medicine Approaches:
- Motor Neuron Replacement:
- Neural stem cell transplantation
- Induced pluripotent stem cell-derived motor neurons
- Challenges in integration and functional connection
- Muscle Regeneration:
- Muscle progenitor cell therapies
- Tissue engineering approaches
- Biomaterial scaffolds for muscle regeneration
- Combination with motor neuron-directed therapies
Prevention Strategies:
- Expanded Genetic Screening:
- Universal carrier screening
- Enhanced prenatal options
- Preimplantation genetic diagnosis accessibility
- Very Early Intervention:
- Pre-symptomatic treatment optimization
- Potential prenatal treatment approaches
- First-trimester screening and intervention
Technological Solutions:
- Advanced Assistive Technology:
- Brain-computer interfaces
- Exoskeleton technology
- Sophisticated mobility solutions
- Environmental control systems
- Functional Electrical Stimulation:
- Targeted muscle activation
- Diaphragmatic pacing
- Neuromuscular electrical stimulation
- Spinal cord stimulation approaches
The research landscape for SMA is exceptionally dynamic, with multiple approaches progressing simultaneously. The field has transitioned from the discovery of disease-modifying therapies to optimizing their use, combining approaches, and developing next-generation treatments. While current therapies have dramatically altered the natural history of SMA, they do not represent complete cures. Future research aims to develop truly curative approaches, further enhance functional outcomes, and address aspects of the disease not fully managed by current therapies. Of particular importance is ensuring these advances are accessible globally, as the gap between treatment possibilities and treatment access remains a significant challenge in many regions.
12. Interesting Facts & Lesser-Known Insights
Uncommon Knowledge About SMA
Genetic Peculiarities:
Unique Evolutionary History: The SMN1/SMN2 duplication is found only in humans, not in other primates or mammals. This relatively recent evolutionary event (approximately 1-2 million years ago) created the unusual situation where humans have two nearly identical SMN genes.
Silent Carrier Phenomenon: Some individuals have two SMN1 copies on one chromosome and none on the other, making them carriers despite having a normal total SMN1 copy number. This “2+0” configuration can lead to unexpected affected children and complicates carrier testing.
SMN Protein Functions: Beyond its well-known role in mRNA splicing, the SMN protein has multiple other functions including roles in:
- DNA damage repair
- Axonal mRNA transport
- Synaptic vesicle release
- Cytoskeletal dynamics
- Mitochondrial function
Genetic Modifiers: Over 30 potential modifier genes have been identified that may influence SMA severity beyond SMN2 copy number, explaining why patients with identical SMN1 and SMN2 genotypes can have different clinical courses.
Clinical Curiosities:
Preserved Cognition: Despite severe motor deficits, cognitive function is preserved and may even be enhanced in some patients with SMA. Some studies suggest slightly above-average verbal IQ in children with SMA.
Differential Vulnerability: Certain motor neuron groups are mysteriously spared in SMA, such as:
- Oculomotor neurons (controlling eye movements)
- Onuf’s nucleus (controlling sphincter muscles)
- Facial nerve nuclei (relatively preserved) This selective vulnerability pattern remains incompletely understood.
Physical Growth Patterns: Children with SMA often have normal or accelerated weight gain despite muscle loss, creating challenges in nutritional management. Some data suggests intrinsic metabolic alterations beyond the effects of decreased activity.
Digital Necrosis: Rarely, infants with severe SMA can develop peripheral vascular complications affecting fingers and toes, potentially related to autonomic dysfunction or microvascular abnormalities.
Reversed Gender Effect: In carriers with SMA symptoms (due to skewed X-inactivation), the disease pattern often differs from typical patients, with more prominent distal rather than proximal weakness.
Myths and Misconceptions
Common Myths vs. Medical Facts:
Myth 1: “SMA affects cognitive development.”
- Fact: Cognitive function is preserved in SMA, with normal to above-average intelligence. Many children with SMA excel academically despite physical limitations.
Myth 2: “All children with SMA Type I die before age 2.”
- Fact: While this reflected the natural history before current treatments, modern care and disease-modifying therapies have dramatically changed outcomes. Many Type I patients now survive well beyond early childhood, and some achieve motor milestones previously thought impossible.
Myth 3: “SMA is a form of muscular dystrophy.”
- Fact: SMA is a motor neuron disease affecting the nerve cells that control muscles, not a primary muscle disease like muscular dystrophy. The muscle weakness in SMA is due to denervation rather than intrinsic muscle pathology.
Myth 4: “SMA progresses at a steady, predictable rate.”
- Fact: The disease often has plateaus and periods of relative stability interspersed with more rapid functional decline. Progression can be quite variable between individuals, even with the same SMN2 copy number.
Myth 5: “Exercise is harmful for patients with SMA.”
- Fact: Appropriate, non-fatiguing exercise can be beneficial for maintaining function and preventing complications. Complete inactivity may accelerate muscle atrophy and contracture development.
Myth 6: “SMA types are fixed categories that determine exact prognosis.”
- Fact: SMA represents a continuous spectrum of severity rather than distinct categories. The traditional classification system provides a useful framework but has limitations, and individuals may not fit perfectly into a single type.
Myth 7: “Carriers of SMA have no symptoms.”
- Fact: While most carriers are asymptomatic, some studies suggest subtle electrophysiological abnormalities or mild motor findings in a subset of carriers. Rarely, female carriers may have significant symptoms due to skewed X-inactivation.
Myth 8: “Gene therapy provides a complete cure for SMA.”
- Fact: While gene therapy can dramatically alter the disease course, current approaches do not fully restore normal function in all patients, particularly when administered after symptom onset. The term “functional cure” may be more appropriate than “complete cure” for current treatments.
Notable Individuals and Cultural Impact
Notable People with SMA:
- Ian Lim: Software engineer at Microsoft and accessibility advocate
- Shane Burcaw: Author, blogger, and disability rights advocate
- Kevan Chandler: Author and world traveler who inspired the “We Carry Kevan” movement
- Mattie Stepanek: Child poet who published several bestselling books before his death at age 13
- Welton Chang: Harvard-educated human rights advocate and policy expert
- Zack Collie: Motivational speaker and disability awareness educator
- Jon Busby: Musician and member of the band Jonsi & Alex
SMA in Media and Culture:
- The documentary “Transfatty Lives” chronicles the life of Patrick O’Brien, a filmmaker with ALS, but raised significant awareness about motor neuron diseases including SMA
- The Netflix documentary “Underwood & Flinch” featured a character with SMA
- The independent film “The Fundamentals of Caring” depicted a teenage character with SMA
- Children’s book “Zoe Bets on Herself” features a protagonist with SMA
- Growing representation in television medical dramas has increased public awareness
Community Innovations:
- Maker Movement Impact: The DIY assistive technology community has created numerous open-source solutions for SMA challenges
- Adaptive Gaming: Significant innovations in accessible gaming have emerged from the SMA community
- Inclusive Fashion: Several fashion lines specifically designed for wheelchair users were inspired by individuals with SMA
- Patient-Led Research: SMA patient advocacy has pioneered models of patient involvement in research design and implementation
Impact on Specific Populations
Geographic and Ethnic Considerations:
- Ashkenazi Jewish Population: Carrier frequency approximately 1 in 41, with specific founder mutations
- Finnish Population: Lower carrier frequency but specific mutations common in Finland
- Spanish Gypsies: Higher prevalence of certain SMA variants
- French-Canadian Population: Founder effect for specific SMN1 mutations in Quebec
- Southeast Asian Populations: Higher frequency of gene conversion events rather than deletions
Family Impact:
- Sibling Experience: Siblings of children with SMA often show remarkable resilience and compassion, but may also experience “well-sibling syndrome” with its own psychological challenges
- Parental Adaptation: Studies show parents of children with SMA develop specialized expertise that often exceeds healthcare professionals in certain aspects of care
- Marital Effects: While some studies suggested higher divorce rates in parents of children with disabilities, more recent research indicates remarkable resilience in many SMA families
- Extended Family Dynamics: Diagnosis often leads to cascade genetic testing throughout extended families, creating complex family dynamics
Educational and Vocational Patterns:
- Despite physical limitations, individuals with SMA often achieve high levels of educational attainment
- Higher than average representation in fields like:
- Computer science and information technology
- Writing and journalism
- Law and advocacy
- Psychology and counseling
- Growing employment opportunities with remote work options
- Pioneering work in accessibility consulting and universal design
Aging with SMA:
- Emerging Phenomenon: The first large cohort of adults with SMA reaching middle and older age
- Unique Challenges:
- Accelerated musculoskeletal aging
- Potential subclinical cardiac involvement emerging late
- Interaction between SMA and age-related conditions
- Reproductive and family planning considerations
- Transition from pediatric to adult care systems
- Resilience Factors: Psychological research identifies remarkable coping strategies and life satisfaction in many adults aging with SMA
Treatment Access Disparities:
- Geographic Disparities: Dramatic differences in treatment access between and within countries
- Socioeconomic Factors: Treatment access often correlates with insurance status and financial resources
- Age-Related Disparities: Treatment approval and coverage often favors pediatric over adult patients
- Advocacy Impact: Patient organization strength significantly influences national treatment access policies
- Ethical Challenges: Allocation of extremely high-cost therapies raises profound ethical questions about resource distribution
The landscape of SMA has been transformed by scientific advances, but many fascinating aspects of the condition remain underappreciated. From its unique genetic features to the remarkable resilience of affected individuals, SMA offers insights into human biology, medicine, and adaptation to physical challenges. The SMA community has been instrumental in driving research forward and developing innovative approaches to living with physical disability, while continuously advocating for both medical advances and social inclusion.
References
Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis. 2017;12(1):124. doi:10.1186/s13023-017-0671-8
Nurputra DK, Lai PS, Harahap NI, et al. Spinal muscular atrophy: from gene discovery to clinical trials. Ann Hum Genet. 2013;77(5):435-463. doi:10.1111/ahg.12031
Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-115. doi:10.1016/j.nmd.2017.11.005
Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018;28(3):197-207. doi:10.1016/j.nmd.2017.11.004
Messina S, Sframeli M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222
Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1723-1732. doi:10.1056/NEJMoa1702752
Mendell JR, Al-Zaidy S, Shell R, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1713-1722. doi:10.1056/NEJMoa1706198
Baranello G, Servais L, Day JW, et al. FIREFISH Part 1: 1-Year Results on Motor Function in Babies with Type 1 Spinal Muscular Atrophy Receiving Risdiplam (RG7916). Neurology. 2019;92(15 Supplement).
Glascock J, Sampson J, Haidet-Phillips A, et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J Neuromuscul Dis. 2018;5(2):145-158. doi:10.3233/JND-180304
Kariyawasam DST, D’Silva A, Lin C, Ryan MM, Farrar MA. Biomarkers and the Development of a Personalized Medicine Approach in Spinal Muscular Atrophy. Front Neurol. 2019;10:898. doi:10.3389/fneur.2019.00898
Dangouloff T, Servais L. Clinical Evidence Supporting Early Treatment of Patients with Spinal Muscular Atrophy: Current Perspectives. Ther Clin Risk Manag. 2019;15:1153-1161. doi:10.2147/TCRM.S172291
Wadman RI, van der Pol WL, Bosboom WM, et al. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2020;1(1):CD006282. doi:10.1002/14651858.CD006282.pub5
Farrar MA, Park SB, Vucic S, et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. 2017;81(3):355-368. doi:10.1002/ana.24864
Calucho M, Bernal S, Alías L, et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28(3):208-215. doi:10.1016/j.nmd.2018.01.003
Pera MC, Coratti G, Forcina N, et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol. 2017;17(1):39. doi:10.1186/s12883-017-0790-9
Hagenacker T, Wurster CD, Günther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020;19(4):317-325. doi:10.1016/S1474-4422(20)30037-5
Kariyawasam D, Carey KA, Jones KJ, Farrar MA. New and developing therapies in spinal muscular atrophy. Paediatr Respir Rev. 2018;28:3-10. doi:10.1016/j.prrv.2018.03.003
Kolb SJ, Coffey CS, Yankey JW, et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. 2017;82(6):883-891. doi:10.1002/ana.25101
Chaytow H, Huang YT, Gillingwater TH, Faller KME. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol Life Sci. 2018;75(21):3877-3894. doi:10.1007/s00018-018-2849-1
Vill K, Kölbel H, Schwartz O, et al. One Year of Newborn Screening for SMA – Results of a German Pilot Project. J Neuromuscul Dis. 2019;6(4):503-515. doi:10.3233/JND-190428
Darras BT, Masson R, Mazurkiewicz-Beldzinska M, et al. Risdiplam-Treated Infants with Type 1 Spinal Muscular Atrophy versus Historical Controls. N Engl J Med. 2021;385(5):427-435. doi:10.1056/NEJMoa2102047
Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med. 2018;378(7):625-635. doi:10.1056/NEJMoa1710504
Day JW, Finkel RS, Chiriboga CA, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20(4):284-293. doi:10.1016/S1474-4422(21)00001-6
Feinberg E, Harvey M, Sheehan P, et al. Parental symptoms of posttraumatic stress after pediatric extracorporeal membrane oxygenation. J Pediatr. 2020;222:91-97. doi:10.1016/j.jpeds.2020.01.073
Deguise MO, Baranello G, Mastella C, et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann Clin Transl Neurol. 2019;6(8):1519-1532. doi:10.1002/acn3.50855
Conclusion
Spinal Muscular Atrophy represents one of the most remarkable success stories in the field of genetic medicine over the past decade. From a devastating neurodegenerative disease with limited treatment options to a condition with multiple FDA-approved therapies, the transformation has been profound. The journey of SMA – from clinical description to genetic understanding to targeted therapy – serves as a model for rare disease research and treatment development.
Several key themes emerge from this comprehensive review:
Early diagnosis and treatment are critical: The implementation of newborn screening and rapid intervention has dramatically altered outcomes, particularly for the most severe forms of SMA.
Multidisciplinary care remains essential: Despite breakthrough therapies, comprehensive supportive care addressing respiratory, nutritional, orthopedic, and psychosocial needs remains fundamental to optimal outcomes.
Treatment landscape continues to evolve: With three distinct treatment approaches now available and numerous others in development, the management paradigm is shifting toward personalized and potentially combination approaches.
Significant challenges remain: Access disparities, high treatment costs, and the incomplete understanding of long-term outcomes represent ongoing challenges in the field.
Patient advocacy has been transformative: The SMA community has driven research forward, influenced policy decisions, and created innovative approaches to living with physical disability.
As we look to the future, the advances in SMA treatment offer hope not only for individuals affected by this condition but also as a blueprint for addressing other genetic disorders. The approach to SMA – combining supportive care with targeted therapies that address the underlying genetic defect – may serve as a model for numerous other conditions. While challenges remain, particularly in ensuring global access to these advances, the progress made in understanding and treating SMA represents one of the most significant success stories in modern medicine.
For patients, families, and healthcare providers navigating the rapidly changing landscape of SMA, maintaining connection with specialized centers, patient advocacy organizations, and the broader SMA community remains essential to accessing the latest advances and optimizing care in this dynamic field.