⚠️ Disclaimer: The information provided in this article is for educational purposes only and does not constitute medical advice. RevisionTown does not provide diagnosis, treatment, or medical recommendations. Always consult a qualified healthcare professional regarding any medical condition, symptoms, or concerns.
Read More – 🏥 Medical Disclaimer
Comprehensive Report on ALS Symptoms
1. Overview
What is ALS?
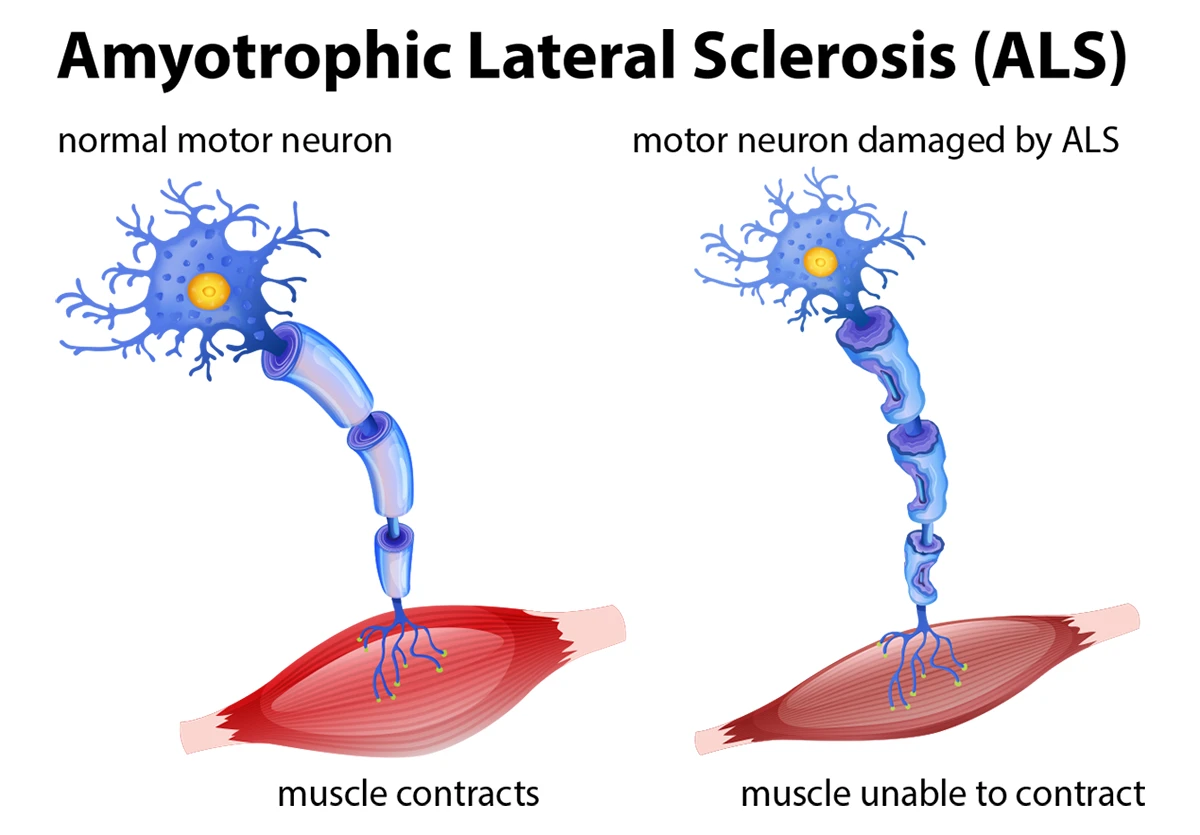
Amyotrophic Lateral Sclerosis (ALS), also known as Lou Gehrig’s disease or Motor Neuron Disease (MND), is a progressive neurodegenerative disorder characterized by the gradual degeneration and death of motor neurons. Motor neurons are nerve cells that control voluntary muscle movement, and their loss leads to muscle weakness, atrophy, and eventually complete paralysis while typically leaving cognitive function intact.
Affected Body Parts/Organs
ALS primarily affects two types of motor neurons:
- Upper motor neurons: Located in the brain’s motor cortex, they send signals to the lower motor neurons
- Lower motor neurons: Located in the brain stem and spinal cord, they directly innervate muscles
As these neurons degenerate, ALS affects virtually all voluntary muscles in the body, including those responsible for:
- Speaking
- Chewing and swallowing
- Breathing
- Movement of limbs
- Trunk control
- Fine motor movements
While ALS primarily affects motor function, some patients may experience cognitive and behavioral changes due to frontotemporal degeneration. The sensory nervous system, bowel, bladder, and sexual function typically remain intact.
Prevalence and Significance
- Global Prevalence: Approximately 5-7 per 100,000 people worldwide
- United States: About 30,000 people currently living with ALS, with roughly 5,000 new diagnoses each year
- Lifetime Risk: About 1 in 300 men and 1 in 400 women will develop ALS
- Age of Onset: Most commonly diagnosed between ages 55-75, though younger-onset cases occur
- Significance: ALS is the most common motor neuron disease in adults and one of the most devastating neurological disorders, with profound impact on quality of life, independence, and survival
2. History & Discoveries
First Identification
ALS was first described in medical literature in 1869 by French neurologist Jean-Martin Charcot, who identified and documented the condition’s distinctive characteristics. Charcot observed both the muscle wasting (amyotrophy) and the hardening of the lateral columns of the spinal cord (lateral sclerosis) in patients, giving the disease its name.
Key Historical Figures
- Jean-Martin Charcot (1825-1893): The “father of neurology” who first described and named the disease “amyotrophic lateral sclerosis”
- Lou Gehrig (1903-1941): The famous American baseball player whose diagnosis in 1939 brought significant public attention to the disease
- Stephen Hawking (1942-2018): Renowned physicist who lived with ALS for 55 years, becoming one of the most famous and longest-surviving patients
Major Discoveries and Breakthroughs
- 1939: ALS gains public awareness after Lou Gehrig’s diagnosis
- 1962: First ALS animal model developed
- 1993: Discovery of the first ALS-linked gene, SOD1, by Robert Brown and colleagues
- 1995: Riluzole becomes the first FDA-approved treatment for ALS
- 2006: TDP-43 protein aggregations identified as a pathological hallmark in most ALS cases
- 2011: Discovery of C9orf72 repeat expansion as the most common genetic cause of ALS
- 2014: Ice Bucket Challenge raises over $115 million for ALS research
- 2017: Edaravone (Radicava) becomes the second FDA-approved drug for ALS
- 2019: First genome-wide association study identifying common genetic risk factors
- 2022: AMX0035 (Relyvrio) receives FDA approval as the third ALS treatment
- 2023-2024: Advanced neuroimaging and biomarker techniques improve diagnosis and prognosis prediction
Evolution of Medical Understanding
Medical understanding of ALS has evolved dramatically:
- Early perspective (1800s-1950s): Considered a purely motor neuron disease with limited understanding of causes
- Mid-century (1950s-1980s): Recognition of clinical variants and patterns of progression
- Genetic era (1990s-2000s): Identification of genetic causes, starting with SOD1 mutations
- Systems perspective (2000s-present): Recognition of ALS as a multisystem disorder involving:
- Protein misfolding and aggregation
- Oxidative stress
- Mitochondrial dysfunction
- RNA processing defects
- Neuroinflammation
- Glutamate excitotoxicity
- Axonal transport impairment
- Overlap with frontotemporal dementia
Modern understanding views ALS as a spectrum disorder with varying presentations, progression rates, and underlying pathologies, rather than a single disease entity.
3. Symptoms
Early Symptoms
Early symptoms of ALS are often subtle and localized, varying based on which motor neurons are affected first:
Limb-Onset ALS (70% of cases):
- Asymmetric weakness in one limb
- Tripping or stumbling
- Difficulty with fine motor tasks (buttoning clothes, writing, using keys)
- Muscle cramps or twitching (fasciculations)
- Muscle stiffness or spasticity
- Hand or foot drop
- Decreased coordination
Bulbar-Onset ALS (25% of cases):
- Slurred speech (dysarthria)
- Difficulty swallowing (dysphagia)
- Excessive saliva (sialorrhea)
- Voice changes (nasal quality, hoarseness)
- Difficulty chewing
- Facial weakness
Truncal/Respiratory-Onset ALS (5% of cases):
- Shortness of breath (dyspnea), especially when lying down
- Decreased vital capacity
- Orthopnea (difficulty breathing when lying flat)
- Reduced exercise tolerance
Advanced-Stage Symptoms
As ALS progresses, symptoms become more widespread and severe:
Motor Function:
- Paralysis of limbs and trunk
- Complete loss of voluntary movement
- Contractures and deformities
- Inability to reposition oneself
- Complete loss of speech
Respiratory Function:
- Respiratory failure
- Inability to clear secretions
- Frequent pneumonia
- Need for mechanical ventilation
Bulbar Function:
- Complete inability to swallow
- Severe dysarthria progressing to anarthria (inability to speak)
- Risk of aspiration
- Need for alternative feeding methods
Autonomic Symptoms (Less Common):
- Excessive sweating
- Temperature dysregulation
- Cardiac arrhythmias
- Blood pressure fluctuations
Common vs. Rare Symptoms
Common Symptoms (Present in >50% of patients):
- Muscle weakness
- Muscle atrophy
- Fasciculations (muscle twitching)
- Spasticity (stiffness)
- Dysarthria (slurred speech)
- Dysphagia (swallowing difficulties)
- Fatigue
- Respiratory insufficiency
- Weight loss
- Cramps
- Emotional lability (pseudobulbar affect)
Uncommon Symptoms (10-50% of patients):
- Cognitive impairment
- Behavioral changes
- Pain (typically secondary to immobility)
- Sleep disturbances
- Depression and anxiety
- Constipation
- Excessive drooling or thick mucus
Rare Symptoms (<10% of patients):
- Sensory abnormalities
- Bowel or bladder dysfunction
- Oculomotor disturbances
- Autonomic dysfunction
- Parkinsonism features
- Pressure sores (despite appropriate care)
- Heart failure (may be part of certain genetic forms)
Symptom Progression Over Time
ALS progression follows a relatively predictable pattern, though speed of progression varies significantly between individuals:
Stage 1 – Early Stage:
- Localized muscle weakness in one region
- Subtle symptoms that may be overlooked or misattributed
- Normal daily functioning with minor adaptations
- Duration: Typically 3-12 months
Stage 2 – Middle Stage:
- Spreading weakness to additional body regions
- Visible muscle atrophy
- Increasing difficulty with daily activities
- Beginning of assistive device use (canes, walkers, braces)
- Duration: Typically 12-24 months
Stage 3 – Advanced Stage:
- Widespread paralysis affecting multiple body regions
- Significant speech and swallowing difficulties
- Respiratory compromise
- Major mobility limitations requiring powered wheelchair
- Duration: Variable, typically 6-12 months
Stage 4 – End Stage:
- Quadriplegia (paralysis of all four limbs)
- Respiratory failure requiring ventilation decisions
- Total dependence for all activities of daily living
- Duration: Variable based on ventilation decisions
Rate of Progression:
- Average survival from symptom onset: 3-5 years
- About 20% of patients survive 5 years
- Approximately 10% survive 10+ years
- More rapid progression typically seen in: older patients, bulbar-onset cases, respiratory-onset cases, and those with associated frontotemporal dementia
- Slower progression more common in: younger patients, certain genetic subtypes (especially SOD1 D90A homozygous), and limb-onset cases
4. Causes
Biological Causes
The precise biological mechanisms causing ALS are complex and likely involve multiple pathways:
Cellular and Molecular Mechanisms:
- Protein Misfolding and Aggregation: Abnormal accumulation of proteins (TDP-43, SOD1, FUS, etc.)
- Oxidative Stress: Excessive free radical production damaging neurons
- Mitochondrial Dysfunction: Impaired energy production in neurons
- Glutamate Excitotoxicity: Overstimulation of neurons leading to cell death
- Impaired Axonal Transport: Disruption of critical cellular cargo movement
- RNA Processing Defects: Abnormal RNA metabolism and processing
- Neuroinflammation: Microglial activation and inflammatory responses
- Autophagy Defects: Impaired cellular waste disposal mechanisms
- Cytoskeletal Abnormalities: Structural problems within motor neurons
- Endoplasmic Reticulum Stress: Problems with protein folding machinery
Genetic and Hereditary Factors
Approximately 5-10% of ALS cases are familial (inherited), while 90-95% are sporadic (no family history). However, genetic factors play a role in both forms:
Major ALS-Linked Genes:
- C9orf72: Hexanucleotide repeat expansion; accounts for 40% of familial and 5-10% of sporadic cases
- SOD1: Superoxide dismutase 1; accounts for 20% of familial and 2% of sporadic cases
- TARDBP: Encodes TDP-43 protein; accounts for 4-5% of familial cases
- FUS: Fused in sarcoma; accounts for 4-5% of familial cases
- Other genes: OPTN, VCP, UBQLN2, TBK1, NEK1, KIF5A, etc.
Genetic Inheritance Patterns:
- Most familial ALS is autosomal dominant (only one affected copy needed)
- Some rare forms show autosomal recessive or X-linked inheritance
- Even in sporadic cases, genetic risk factors contribute to susceptibility
Environmental Causes and Risk Factors
While no single environmental factor has been definitively proven to cause ALS, several have been associated with increased risk:
Established Environmental Risk Factors:
- Military Service: Particularly Gulf War veterans
- Repeated Head Trauma: Professional athletes in contact sports
- Certain Occupations: Agricultural work, electrical work, welding
Possible Environmental Risk Factors:
- Heavy Metal Exposure: Lead, mercury, manganese, selenium
- Pesticide/Herbicide Exposure: Agricultural chemicals
- Electromagnetic Field Exposure: High-voltage power lines
- Cyanobacteria Exposure: Certain harmful algal blooms
Known Triggers or Exposure Risks
Some factors may trigger disease onset in genetically susceptible individuals:
- Physical Trauma: Intense physical activity or injury possibly triggering onset in predisposed individuals
- Viral Infections: Certain viral infections may trigger inflammatory responses
- Metabolic Stress: Periods of severe metabolic disturbance
- Toxic Exposures: Acute exposure to certain neurotoxins
- Aging: Age-related cellular decline as the most consistent risk factor
Current consensus suggests ALS likely results from a complex interplay between genetic predisposition and environmental exposures, following a “multiple hit” hypothesis rather than having a single cause.
5. Risk Factors
Demographic Risk Factors
Age:
- Peak age of onset: 55-75 years
- Median age at diagnosis: 64-67 years
- Juvenile onset (<25 years): Very rare (<1% of cases)
- Risk increases with age until around 75, then decreases
Gender:
- Men have slightly higher risk than women (1.5:1 ratio)
- This gender difference decreases with advancing age
- Bulbar-onset more common in women
- Post-menopausal women show increased incidence, suggesting possible hormonal influences
Race/Ethnicity:
- Slightly higher rates in Caucasian populations
- Lower rates in Hispanic, Asian, and African populations
- Specific genetic subtypes vary by ethnicity (e.g., different SOD1 mutations)
- Mortality rates vary significantly between countries
Geographic Factors:
- Western Pacific ALS hotspots (Guam, Kii Peninsula, West Papua)
- North-to-south gradient in Europe (higher rates in northern countries)
- Higher rates in certain US regions (Northeast)
Environmental and Occupational Risk Factors
Environmental Exposures:
- Agricultural Chemicals: Pesticides, herbicides, fungicides
- Heavy Metals: Lead, mercury, arsenic, cadmium, selenium
- Solvents: Industrial cleaning agents, degreasers
- Cyanobacteria: Beta-methylamino-L-alanine (BMAA) toxin exposure
Occupational Risk Factors:
- Military Service: 1.5-2 times increased risk in veterans, particularly Gulf War veterans
- Agricultural Work: Farmers and agricultural workers
- Construction Work: Possibly due to chemical exposures
- Electrical Work: Electricians and power-line workers
- Professional Sports: Football, soccer, ice hockey (head trauma)
Lifestyle Factors:
- Smoking: 1.4-1.6 times increased risk, dose-dependent
- Intense Physical Activity: Professional athletes and intense recreational exercise
- Diet: Possible associations with low antioxidant intake
- Body Mass Index: Lower premorbid BMI associated with increased risk
Genetic Risk Factors
Familial Risk:
- 5-10% of ALS is familial (having affected family members)
- First-degree relatives of ALS patients have 4-8 times increased risk
- Over 50 genes identified with varying effects on risk
Known Genetic Risk Modifiers:
- UNC13A variants: Affect progression rate
- ATXN2 intermediate expansions: Increase susceptibility
- EPHA4 variants: Modify disease onset and progression
- Apolipoprotein E (APOE): ε4 allele may increase risk
Ancestry-Specific Genetic Factors:
- Different genetic mutations predominate in different populations
- Lower C9orf72 frequency in Asian populations
- Higher SOD1 mutations in Scandinavian populations
Impact of Pre-existing Conditions
Conditions Associated with Increased ALS Risk:
- Frontotemporal Dementia: Strong bidirectional relationship with ALS
- Autoimmune Disorders: Small increased risk with some autoimmune conditions
- Traumatic Brain Injury: Modest association, particularly with repeated injuries
- Metabolic Disorders: Diabetes may alter disease course
- Viral Infections: HIV, polio, other neurotropic viruses (controversial)
Protective Associations:
- Hyperlipidemia: Paradoxically associated with better prognosis
- Obesity: Higher BMI associated with better survival after diagnosis
- Cancer History: Possible inverse relationship between cancer and ALS
6. Complications
Direct Complications of ALS
Respiratory Complications:
- Respiratory Failure: Primary cause of death in 70% of ALS patients
- Aspiration Pneumonia: Due to impaired swallowing and cough
- Sleep-Disordered Breathing: Sleep apnea, hypopnea, hypoxemia
- Reduced Vital Capacity: Declining ability to inhale deeply and exhale forcefully
- Atelectasis: Lung collapse due to inability to fully expand lungs
- Mucus Plugging: Inability to clear secretions
Nutritional Complications:
- Malnutrition: Due to dysphagia and increased metabolic demands
- Dehydration: Difficulty maintaining adequate fluid intake
- Weight Loss: Accelerated loss of lean body mass
- Hypermetabolism: Increased caloric needs despite reduced activity
Musculoskeletal Complications:
- Contractures: Permanent joint tightening due to immobility
- Pressure Injuries: Skin breakdown due to inability to shift position
- Fractures: Due to falls and osteopenia from immobility
- Deep Vein Thrombosis: Blood clots from immobility
- Pulmonary Embolism: Life-threatening complication of DVT
Neurological Complications:
- Pseudobulbar Affect: Inappropriate emotional outbursts
- Cognitive Impairment: Frontotemporal spectrum disorders in 30-50%
- Depression and Anxiety: Reactive and neurobiological
- Pain Syndromes: Musculoskeletal pain, cramps, spasticity
Long-term Impact on Health
Physical Impacts:
- Progressive loss of independence in all activities of daily living
- Complete paralysis in advanced stages
- Need for 24-hour care
- Reliance on assistive technology for communication and mobility
- Ventilator dependence in later stages (if chosen)
Psychological Impacts:
- Existential distress
- Loss of autonomy
- Changes in family dynamics and roles
- Social isolation
- Grief over progressive losses
Quality of Life Impacts:
- Decreasing mobility and independence
- Communication challenges
- Feeding difficulties
- Respiratory distress
- Financial burden
- Caregiver strain
Disability and Fatality Rates
Disability Progression:
- Average time from diagnosis to wheelchair use: 12-18 months
- Average time from diagnosis to significant speech impairment: 14-22 months
- Average time from diagnosis to requiring feeding tube: 18-24 months
- Average time from diagnosis to requiring ventilation: 24-30 months
Survival Statistics:
- Median survival from symptom onset: 3-5 years
- 20% of patients survive 5 years or longer
- 10% of patients survive 10 years or longer
- 2-5% survive 20 years or longer
- Median survival with tracheostomy ventilation: 6-10 years
- Median survival with non-invasive ventilation only: 7-15 months after initiation
Prognostic Factors Affecting Survival:
- Age at onset (younger = longer survival)
- Site of onset (limb onset = better prognosis than bulbar or respiratory)
- Rate of progression before diagnosis
- Early respiratory involvement (poor prognostic sign)
- Presence of cognitive impairment (reduces survival)
- Access to multidisciplinary care (improves outcomes)
- Nutritional status
7. Diagnosis & Testing
Common Diagnostic Procedures
Clinical Evaluation:
- Comprehensive neurological examination
- Detailed medical history
- Review of symptom progression
- Family history assessment
- Physical assessment of muscle strength, tone, and reflexes
El Escorial Criteria: Internationally recognized diagnostic criteria that classify ALS cases as:
- Definite ALS: Upper and lower motor neuron signs in at least three regions
- Probable ALS: Upper and lower motor neuron signs in at least two regions
- Possible ALS: Upper and lower motor neuron signs in one region
- Suspected ALS: Lower motor neuron signs only in two or more regions
Awaji Criteria: Modified criteria that incorporate electrophysiological findings as equivalent to clinical findings, increasing diagnostic sensitivity.
Medical Tests
Electrophysiological Studies:
- Electromyography (EMG): Measures electrical activity in muscles, detecting denervation signs
- Nerve Conduction Studies (NCS): Assesses nerve function to rule out other disorders
- Motor Unit Number Estimation (MUNE): Quantifies surviving motor neurons
- Transcranial Magnetic Stimulation (TMS): Assesses upper motor neuron function
Laboratory Tests:
- Blood Tests: To exclude mimicking conditions
- Complete blood count
- Comprehensive metabolic panel
- Thyroid function tests
- Creatine kinase levels
- Vitamin B12 and folate levels
- Inflammatory markers
- Paraneoplastic antibody panel
- Heavy metal screening
- Ganglioside antibodies
- Anti-MAG antibodies
- Cerebrospinal Fluid Analysis: May be performed to rule out inflammatory conditions
- Genetic Testing: For SOD1, C9orf72, FUS, TARDBP, and other ALS-linked genes
Imaging Studies:
- MRI of Brain and Spinal Cord: To exclude structural lesions and observe neurodegeneration
- Diffusion Tensor Imaging (DTI): Advanced MRI technique to visualize white matter tracts
- SPECT/PET Scanning: Functional imaging showing metabolic changes
- Muscle Ultrasound: Can show fasciculations and muscle thinning
Specialized Testing:
- Muscle Biopsy: Occasionally performed to rule out myopathies
- Respiratory Function Tests:
- Forced vital capacity (FVC)
- Maximal inspiratory pressure (MIP)
- Sniff nasal inspiratory pressure (SNIP)
- Nocturnal oximetry
- Swallowing Studies: Videofluoroscopic swallowing examination
- Neuropsychological Testing: To assess cognitive function
Early Detection Methods and Effectiveness
Current Challenges in Early Detection:
- Average diagnostic delay: 10-16 months from symptom onset
- Initial misdiagnosis rate: 25-40%
- No definitive diagnostic biomarker yet available
- Early symptoms often subtle and non-specific
Emerging Biomarkers:
- Neurofilament Light Chain (NfL): Blood and CSF biomarker showing promise
- Sensitivity: 85-90%
- Specificity: 70-85%
- MicroRNAs: Specific patterns of circulating microRNAs
- Proteomics: Multi-protein panels showing diagnostic potential
- Neuroimaging Biomarkers: Advanced MRI techniques showing characteristic patterns
- Skin Biopsy: TDP-43 pathology detection in skin nerve fibers
Effectiveness of Early Diagnosis:
- Earlier intervention with riluzole may improve survival by 2-3 months
- Earlier nutritional intervention prevents malnutrition
- Earlier respiratory support improves quality of life
- Time to access clinical trials and experimental treatments
- Psychological preparation and life planning
- However, still no disease-modifying intervention that dramatically alters course if started earlier
8. Treatment Options
Standard Treatment Protocols
Multidisciplinary Care Approach: The cornerstone of ALS management is comprehensive care through a multidisciplinary team typically including:
- Neurologist (preferably with ALS specialization)
- Respiratory therapist
- Physical therapist
- Occupational therapist
- Speech-language pathologist
- Nutritionist
- Social worker
- Palliative care specialist
- ALS nurse coordinator
Studies show multidisciplinary clinic attendance improves survival by 7-12 months compared to general neurology care.
Medications
FDA-Approved Disease-Modifying Treatments:
- Riluzole (Rilutek): Anti-glutamatergic medication
- Mechanism: Reduces glutamate excitotoxicity
- Efficacy: Extends survival by 2-3 months on average
- Dosage: 50mg twice daily
- Side effects: Fatigue, nausea, elevated liver enzymes
- Edaravone (Radicava): Free radical scavenger
- Mechanism: Reduces oxidative stress
- Efficacy: Slows functional decline by approximately 33% in a subset of patients
- Administration: Intravenous infusion or oral formulation
- Side effects: Bruising, gait disturbances, headache
- AMX0035 (Relyvrio): Combination of sodium phenylbutyrate and taurursodiol
- Mechanism: Reduces endoplasmic reticulum stress and mitochondrial dysfunction
- Efficacy: Extended median survival by approximately 6.5 months in clinical trials
- Administration: Oral suspension
- Side effects: Diarrhea, abdominal pain, nausea
Symptomatic Medications:
- For Spasticity: Baclofen, tizanidine, dantrolene, botulinum toxin injections
- For Cramps: Quinine, magnesium, mexiletine
- For Excess Saliva: Anticholinergics, botulinum toxin injections to salivary glands
- For Pseudobulbar Affect: Dextromethorphan/quinidine (Nuedexta)
- For Pain: NSAIDs, gabapentin, pregabalin
- For Depression/Anxiety: SSRIs, SNRIs, benzodiazepines
- For Insomnia: Melatonin, zolpidem, trazodone
- For Fasciculations: Membrane stabilizers (limited effectiveness)
Supportive and Adaptive Therapies
Respiratory Support:
- Non-invasive Ventilation (NIV): BiPAP or similar devices
- Extends survival by 7-12 months on average
- Improves quality of life and sleep
- Typically initiated when FVC falls below 50%
- Invasive Ventilation: Tracheostomy with mechanical ventilation
- Can extend survival for many years
- Requires 24/7 caregiving resources
- Significant ethical and quality-of-life considerations
- Cough Assist Devices: Mechanical insufflation-exsufflation
- Suction Machines: For secretion management
Nutritional Support:
- Percutaneous Endoscopic Gastrostomy (PEG): Feeding tube
- Recommended when weight loss exceeds 10% or significant dysphagia
- Extends survival by 3-6 months on average
- Can still enjoy oral feeding for pleasure if safe
- Dietary Modifications: High-calorie, easy-to-swallow foods
- Nutritional Supplements: Ensure adequate caloric intake
- Hydration Management: Thickened liquids, adequate fluid intake
Rehabilitative Therapies:
- Physical Therapy: Range of motion exercises, strength maintenance
- Occupational Therapy: Adaptive equipment, energy conservation
- Speech Therapy: Communication strategies, AAC devices
- Respiratory Therapy: Breathing exercises, airway clearance
- Swallowing Therapy: Safe swallowing techniques
Assistive Devices:
- Mobility Aids: Ankle-foot orthoses, walkers, wheelchairs
- Communication Devices: Eye-tracking systems, voice banking
- Environmental Controls: Home automation systems
- Adaptive Equipment: Modified utensils, dressing aids
Emerging Treatments and Clinical Trials
Promising Therapeutic Approaches:
- Gene Therapy: Antisense oligonucleotides targeting SOD1, C9orf72, and other genes
- Stem Cell Therapy: Mesenchymal and neural stem cell transplantation
- Immunomodulation: Targeting neuroinflammation with monoclonal antibodies
- Small Molecules: Targeting various pathways (protein misfolding, autophagy, etc.)
- Repurposed Medications: Existing drugs with potential neuroprotective effects
Notable Clinical Trials (as of 2025):
- Tofersen: Antisense oligonucleotide for SOD1-ALS
- Masitinib: Tyrosine kinase inhibitor with anti-inflammatory properties
- Verdiperstat: Myeloperoxidase inhibitor targeting neuroinflammation
- NurOwn: Mesenchymal stem cell therapy
- AT-1501: Anti-CD40L antibody for immunomodulation
- CNM-Au8: Gold nanocrystal suspension targeting cellular energetics
- IONIS-C9Rx: Antisense oligonucleotide for C9orf72-ALS
For current trial information, patients should consult clinicaltrials.gov or ALS research foundations, as the landscape changes rapidly.
9. Prevention & Precautionary Measures
Primary Prevention
Currently, there are no proven methods to prevent ALS in the general population. However, some strategies may potentially reduce risk based on epidemiological evidence:
Potential Preventive Measures:
- Antioxidant-Rich Diet: High consumption of colorful fruits and vegetables
- Omega-3 Fatty Acids: Regular consumption of fatty fish or supplements
- Vitamin E: Foods rich in vitamin E or supplementation
- Regular Physical Activity: Moderate (not excessive) exercise
- Avoiding Environmental Toxins: Minimizing exposure to pesticides, heavy metals
- Smoking Cessation: Reducing a known risk factor
- Head Trauma Prevention: Avoiding high-impact sports or using proper protection
Lifestyle Recommendations for At-Risk Individuals
For those with familial ALS or genetic predisposition:
- Regular Neurological Check-ups: Early detection of subtle symptoms
- Genetic Counseling: For family planning decisions
- Stress Management: As stress may potentially trigger onset in susceptible individuals
- Nutritional Optimization: Balanced diet rich in antioxidants
- Weight Maintenance: Mild overweight status may be protective
- Moderate Exercise: Avoiding extreme physical exertion
- Occupational Considerations: Avoiding high-risk occupations
Secondary Prevention for Diagnosed Patients
For patients already diagnosed with ALS, measures to prevent complications include:
Respiratory Complications Prevention:
- Early NIV initiation
- Regular respiratory function monitoring
- Pneumococcal and influenza vaccinations
- Proper positioning and postural drainage
Nutritional Complications Prevention:
- Early nutritional intervention
- PEG placement before severe weight loss
- Regular monitoring of weight and nutritional status
- High-calorie, high-protein diet
Musculoskeletal Complications Prevention:
- Regular range of motion exercises
- Proper positioning and frequent repositioning
- Pressure-relieving mattresses and cushions
- Ankle-foot orthoses to prevent contractures
Psychological Complications Prevention:
- Early psychological support
- Support groups participation
- Proactive discussion of disease progression and planning
- Attention to quality of life concerns
Predictive Testing and Screening
For familial ALS, genetic testing options include:
- Diagnostic Testing: For symptomatic individuals
- Predictive Testing: For asymptomatic individuals with family history
- Prenatal Testing: For families with known genetic mutations
- Carrier Testing: For recessive forms of ALS
These testing options come with significant ethical considerations and should be accompanied by genetic counseling.
10. Global & Regional Statistics
Global Incidence and Prevalence
Global Incidence:
- Worldwide incidence: 1-2.6 per 100,000 person-years
- Approximately 223,000 new cases diagnosed annually worldwide
- Estimated 5-7 people per 100,000 living with ALS globally
Regional Variations in Incidence:
- Europe: 2-3 per 100,000 person-years
- North America: 1.5-2.5 per 100,000 person-years
- Asia: 0.7-1.5 per 100,000 person-years
- South America: 0.3-1.5 per 100,000 person-years
- Africa: Limited data, estimated 0.5-1 per 100,000 person-years
Prevalence Rates:
- Global prevalence: 4.1-8.4 per 100,000 population
- North America: 3.4-7.9 per 100,000
- Europe: 5.4-7.3 per 100,000
- Asia: 1-5 per 100,000
- Oceania: 2.1-5.7 per 100,000
Mortality and Survival Rates
Global Mortality:
- ALS causes approximately 69,000 deaths worldwide annually
- Mortality rates closely track incidence rates due to the disease’s fatal nature
Survival Rates:
- Median survival from diagnosis: 20-48 months
- 5-year survival rate: 20%
- 10-year survival rate: 10%
- 20-year survival rate: 2-5%
Factors Affecting Survival:
- Highest survival rates in countries with:
- Universal healthcare systems
- Access to multidisciplinary ALS clinics
- Early diagnosis
- Higher socioeconomic status
- Greater availability of assistive technology and home care
Country-wise Comparison and Trends
Highest ALS Rates:
- Finland: 2.4 per 100,000 person-years
- United States: 2.2 per 100,000 person-years
- Sweden: 2.2 per 100,000 person-years
- Scotland: 2.1 per 100,000 person-years
- Ireland: 2.1 per 100,000 person-years
Lowest ALS Rates:
- China: 0.3-0.8 per 100,000 person-years
- Iran: 0.4 per 100,000 person-years
- Mexico: 0.4 per 100,000 person-years
- South Africa: Limited data, estimated 0.3-0.6 per 100,000 person-years
ALS Hotspots:
- Kii Peninsula, Japan
- Guam (declining since the 1950s)
- Parts of Western New Guinea
- Sardinia, Italy
Global Trends:
- Rising Incidence: 32% increase globally between 1990-2020
- Aging Population Effect: Increasing cases due to global aging
- Diagnostic Improvements: Better recognition leading to higher rates
- Closing Gender Gap: Male:female ratio decreasing over time
- Changing Geographic Patterns: Rising rates in developing countries
11. Recent Research & Future Prospects
Latest Advancements in Treatment and Research
Therapeutic Advancements:
- Antisense Oligonucleotides (ASOs): Tofersen for SOD1-ALS showing promising results in slowing progression
- Gene Therapy Approaches: AAV-delivered gene therapies advancing to clinical trials
- Small Molecule Drug Development: Targeted approaches to specific cellular mechanisms
- Precision Medicine: Biomarker-guided treatment selection
- Combination Therapies: Multiple drugs targeting different pathways simultaneously
- Improved Assistive Technology: Brain-computer interfaces for communication
Diagnostic Advancements:
- Liquid Biomarkers: Blood and CSF biomarkers (neurofilament light chain, TDP-43, microRNAs)
- Advanced Neuroimaging: Multimodal MRI protocols for earlier detection
- Electrical Impedance Myography: Non-invasive muscle assessment
- Exhaled Breath Analysis: Volatile compound detection
- Digital Phenotyping: Smartphone-based symptom monitoring
- Voice Analysis: AI detection of subtle speech changes
Research Methodologies:
- ALS Patient-Derived iPSCs: Creating personalized cellular models
- CRISPR Gene Editing: Creating precise disease models and potential therapies
- Machine Learning Algorithms: For prognosis prediction and subtype classification
- Wearable Technology: Continuous monitoring of disease progression
- Platform Trials: Testing multiple therapies simultaneously with shared placebo groups
Ongoing Studies and Future Medical Possibilities
Major Research Directions:
- Genetic Modifiers: Identifying factors that influence penetrance and progression
- Gut Microbiome Studies: Exploring gut-brain axis in ALS
- Environmental Exposome Mapping: Comprehensive measurement of environmental exposures
- Neuroprotective Strategies: Preserving motor neuron function
- Neuroinflammation Modulation: Targeting microglia and astrocytes
- RNA Metabolism: Targeting RNA processing defects
- Protein Homeostasis: Addressing protein misfolding and aggregation
- Axonal Transport Enhancement: Improving cellular cargo movement
- Metabolic Dysfunction: Targeting cellular energetics
Future Medical Approaches:
- Combinatorial Therapies: Multiple agents targeting complementary pathways
- Personalized Medicine: Treatment selection based on genetic and biomarker profiles
- Extended-Release Formulations: Improving drug delivery and compliance
- Nanotechnology-Based Delivery: Targeted drug delivery to CNS
- Remote Monitoring Systems: Telemedicine approaches to care
Potential Cures or Innovative Therapies Under Development
While a complete cure remains elusive, several approaches show potential for significant disease modification:
Gene-Targeted Approaches:
- ASO Therapies: Targeting specific genetic mutations
- Gene Replacement: For loss-of-function mutations
- Gene Editing: CRISPR-based approaches for mutation correction
- RNA-Targeted Therapies: Small molecules affecting RNA processing
Cell-Based Therapies:
- Stem Cell Transplantation: NurOwn, CNS10-NPC, and other cell therapies
- Cell-Based Growth Factor Delivery: Engineered cells producing neurotrophic factors
- Immunomodulatory Cell Therapies: Regulatory T-cells to dampen neuroinflammation
Innovative Delivery Systems:
- Blood-Brain Barrier Penetration Technologies: Enhanced CNS drug delivery
- Intrathecal Drug Delivery Systems: Direct delivery to cerebrospinal fluid
- Viral Vector Delivery: AAV-based gene therapy delivery
Disease-Modifying Combinations:
- Multi-Drug Regimens: Combining existing and new therapies
- Pathway-Specific Combinations: Targeting multiple aspects of a single pathway
- Stage-Specific Approaches: Different treatments at different disease stages
12. Interesting Facts & Lesser-Known Insights
Uncommon Knowledge About ALS
Historical Facts:
- First Description: While Charcot is credited with first describing ALS in 1869, similar descriptions date back to ancient Egyptian medical papyri
- Guam Phenomenon: After WWII, up to 1 in 3 deaths among Chamorro people of Guam were due to ALS, leading to the discovery of cycad toxin exposure as a potential trigger
- Lou Gehrig May Not Have Had ALS: Some researchers suggest he might have suffered from chronic traumatic encephalopathy (CTE) due to concussions
Scientific Curiosities:
- Eye Movements Preserved: Even in late-stage ALS, external eye muscles remain functional, allowing for eye-tracking communication
- Sensory Neurons Spared: ALS typically spares sensory function, though subtle sensory changes are increasingly recognized
- Bladder/Bowel Control Maintained: Autonomic neurons controlling these functions are typically preserved
- Cancer Inverse Relationship: Patients with ALS have lower rates of cancer, suggesting potentially related but opposing cellular mechanisms
- “ALS Personality”: Some studies suggest personality traits like perfectionism and high achievement may be more common in ALS patients
Demographic Patterns:
- “Old” Versus “Young” ALS: Different ages of onset are associated with different genetic causes and disease courses
- Marital Status Effect: Married patients tend to live longer after diagnosis, likely due to caregiving support
- Handedness: Left-handed people may have slightly higher ALS risk
- Athleticism Paradox: Regular exercise is protective, but elite athletics may increase risk
Myths and Misconceptions vs. Medical Facts
Myth: ALS affects cognitive function in most patients. Fact: While cognitive changes occur in approximately 50% of patients, they are often subtle, and only 15-20% develop frank dementia.
Myth: ALS is strictly a motor neuron disease with no other components. Fact: Modern research shows ALS affects multiple cellular systems and has overlap with frontotemporal dementia.
Myth: Only old people get ALS. Fact: While most common in people 55-75, ALS can strike at any adult age, with about 10% of cases occurring before age 40.
Myth: ALS is contagious. Fact: ALS cannot be transmitted from person to person through any known mechanism.
Myth: ALS progression is the same for everyone. Fact: The disease course varies dramatically between individuals, with some progressing rapidly over months while others live decades.
Myth: All ALS patients eventually need ventilators. Fact: While respiratory compromise is common, not all patients choose ventilation, and some die from other complications.
Myth: ALS patients are completely locked in their bodies but mentally sharp. Fact: While many maintain cognitive function, up to 50% experience some cognitive or behavioral changes.
Myth: People with ALS can’t feel anything physically. Fact: Sensory function typically remains intact, and pain from immobility, cramps, and spasticity is common.
Impact on Specific Populations and Professions
Professional Athletes:
- Higher rates observed in Italian soccer players (6x higher)
- NFL players have 4x higher risk of dying from ALS
- Potential mechanisms include head trauma, extreme physical exertion, performance-enhancing drugs
Military Veterans:
- 1.5-2x higher risk, particularly Gulf War veterans
- Possible contributors include environmental exposures, trauma, and stress
- Veterans may qualify for presumptive service connection benefits
Young-Onset ALS Patients (under 40):
- More likely to have genetic forms
- Often slower progression
- Higher likelihood of upper limb onset
- Greater burden on career, young families
- Specialized needs related to fertility and family planning
Elderly-Onset ALS Patients (over 75):
- Often more rapid progression
- More likely to have bulbar onset
- Higher comorbidity burden
- Treatment tolerance challenges
- End-of-life planning often more accepting
Working Professionals:
- Average age at diagnosis coincides with peak career years
- Workplace challenges including disability disclosure
- Requirements for workspace adaptations
- Challenges with health insurance and disability benefits
- Career termination typically precedes major care needs
ALS Caregivers:
- Extraordinary burden: average 11-15 hours daily care in advanced stages
- Higher rates of depression, anxiety, and burnout
- Financial strain from reduced work hours and direct care costs
- 11% of family caregivers report health deterioration
- Positive aspects include deepened relationships and personal growth
This comprehensive report on ALS symptoms has been compiled from current medical literature and research through October 2024. While extensive, it should be used for informational purposes only and not as a substitute for professional medical advice.